
Abstract
Age is the dominant risk factor for infectious diseases, but the mechanisms linking age to infectious disease risk are incompletely understood. Age-related mosaic chromosomal alterations (mCAs) detected from genotyping of blood-derived DNA, are structural somatic variants indicative of clonal hematopoiesis, and are associated with aberrant leukocyte cell counts, hematological malignancy, and mortality. Here, we show that mCAs predispose to diverse types of infections. We analyzed mCAs from 768,762 individuals without hematological cancer at the time of DNA acquisition across five biobanks. Expanded autosomal mCAs were associated with diverse incident infections (hazard ratio (HR) 1.25; 95% confidence interval (CI) = 1.15–1.36; P = 1.8 × 10−7), including sepsis (HR 2.68; 95% CI = 2.25–3.19; P = 3.1 × 10−28), pneumonia (HR 1.76; 95% CI = 1.53–2.03; P = 2.3 × 10−15), digestive system infections (HR 1.51; 95% CI = 1.32–1.73; P = 2.2 × 10−9) and genitourinary infections (HR 1.25; 95% CI = 1.11–1.41; P = 3.7 × 10−4). A genome-wide association study of expanded mCAs identified 63 loci, which were enriched at transcriptional regulatory sites for immune cells. These results suggest that mCAs are a marker of impaired immunity and confer increased predisposition to infections.
Similar content being viewed by others

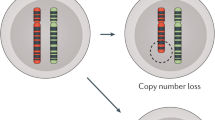

Main
With advancing age comes increased susceptibility to infectious diseases1,2. Immunosenescence is the age-related erosion of immune function, particularly with respect to adaptive immunity3,4,5,6. Leukocytes, including T cells and B cells, are key mediators of adaptive host defenses against infections, with impaired immune responses increasing the risk for infections7,8,9. Age-related mosaic chromosomal alterations (mCAs) detected from blood-derived DNA are clonal structural somatic alterations (deletions, duplications, or copy number neutral loss of heterozygosity (CNN-LOH)) present in a fraction of peripheral leukocytes that can indicate clonal hematopoiesis10,11,12. mCAs are associated with aberrant leukocyte cell count and an increased risk for hematological malignancy and mortality10,11,12,13,14,15,16,17,18.
Although the relationship between mCAs and increased hematologic cancer risk is well-established10,11,12, the impact of mCAs on age-related diminishment of immune function is poorly understood. We propose that mCAs increase the risk of infection, given that mCAs are somatic variants that increase in abundance with age and are associated with alterations in leukocyte count. In this study, we harness DNA genotyping array intensity data and long-range chromosomal phase information inferred from 768,762 individuals across five biobanks to analyze the associations between expanded mCA clones (that is, mCAs present in at least 10% of peripheral leukocyte DNA indicative of clonal expansion) and diverse infections, including severe coronavirus disease 2019 (COVID-19) from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection (Fig. 1a). To elucidate genetic risk factors for the development of expanded mCA clones, we performed a genome-wide association study (GWAS) of data from the UK Biobank and subsequent in silico cell-specific, transcriptomic, and pathway analyses.

a, Genome-wide mCAs were detected across the UKB11, MGBB (via the MoChA pipeline, https://github.com/freeseek/mocha), FinnGen (via the MoChA pipeline), BBJ10 and CUB (via the MoChA pipeline) cohorts. The association of expanded mCAs (cell fraction > 10%) with incident infectious diseases in the UKB, MGBB, and FinnGen cohorts, with incident infectious disease mortality in the BBJ cohort, and with COVID-19 severity in COVID-19-positive cases in the CUB, was assessed. A GWAS for expanded mCAs was then performed in the UKB to identify causal factors for expanded mCAs. Using the GWAS results, cell-specific functional enrichment analyses were performed using GenoSkyline-Plus, which combines epigenetic and transcriptomic annotations with GWAS summary statistics to estimate the relative contribution of cell-specific functional markers to the GWAS results. Additionally, to prioritize putative causal genes and pathways promoting the development of expanded mCAs, whole-blood TWAS was performed using UTMOST via GTExv8. b,c, The association of all expanded mCAs (cell fraction > 10%) (b) and expanded autosomal mCAs (cell fraction > 10%) (c) with age, stratified by sex for individuals in the UKB, MGBB, FinnGen and BBJ cohorts combined. Error bands are derived from binomial proportion 95% CIs. Plots by cohort and across other mCA groupings are available in Supplementary Figs. 8 and 9. BBJ, BioBank Japan; CUB, Columbia University Biobank; GTEx v8, Genotype-Tissue Expression Project version 8; GWAS, genome-wide association study; MGBB, Mass General Brigham Biobank; mCA, mosaic chromosomal alterations; MoChA, Mosaic Chromosomal Alterations software (https://github.com/freeseek/mocha); TWAS, transcriptome-wide association study; UKB, UK Biobank; UTMOST, Unified Test for Molecular Signatures.
Results
Population characteristics and mCA prevalence
Data from a total of 768,762 unrelated, multi-ethnic individuals from the UK Biobank (UKB) (n = 444,199), Mass General Brigham Biobank (MGBB) (n = 22,461), FinnGen cohort (n = 175,690), BioBank Japan (BBJ) (n = 125,541) and the Columbia University Biobank (CUB) (n = 871) who passed genotype and mCA quality control criteria (Supplementary Figs. 1–7) were analyzed (Supplementary Table 1). The mCA calls from the UKB and BBJ are taken from studies that have been performed previously10,11, while the Mosaic Chromosomal Alterations (MoChA) pipeline (https://github.com/freeseek/mocha) was used to detect mCAs in the MGBB, FinnGen, and CUB cohorts (Extended Data Fig. 1) from genome-wide genotyping of blood DNA. For the UKB participants, the mean age at DNA collection was 57 (s.d. 8) years, 204,579 (46.1%) were male, 188,875 (45.0%) were prior or current smokers, and 66,551 (15.0%) had a history of solid cancer. In the MGBB participants, the mean age was 55 (s.d. 17) years, 10,306 (45.9%) were male, 9,094 (40.5%) were prior or current smokers, and 6,080 (27.1%) had a history of solid cancer. In the FinnGen participants, the mean age was 53 (s.d. 18) years, 71,000 (40.4%) were male, 42.7% were prior or current smokers (when smoking status was available) and 31,855 (18.1%) had a history of solid cancer. In the BBJ participants, the mean age was 65 (s.d. 12) years, 72,186 (57.5%) were male, and 66,913 (53.3%) were prior or current smokers, and 25,987 (20.7%) had a history of solid cancer. In the CUB participants, the mean age was 62.3 (s.d. 17.9) years, 480 (55.1%) were male, and 221 (25.4%) had a history of solid cancer (Supplementary Table 1).
In the UKB cohort, of 444,199 unrelated individuals without a known history of hematologic malignancy, 66,011 (14.9%) carried an mCA (15,350 autosomal) and 12,398 (3.2%) carried an expanded mCA clone, defined as an mCA mutation present in at least 10% of peripheral leukocytes (2,985 autosomal) (Supplementary Table 2). Although most of the carriers had only one mCA, 6% of individuals carried between 2 and 22 non-overlapping mCAs (Supplementary Fig. 7). In the MGBB cohort, of 22,461 unrelated individuals without a history of hematologic cancer, 3,784 (16.8%) carried an mCA (1,025 autosomal) and 1,026 (5.2%) carried an expanded mCA clone (337 autosomal). In the FinnGen cohort, of 175,690 individuals without a history of hematologic cancer, 22,040 (12.5%) carried an mCA (3,164 autosomal) and 9,558 (5.9%) carried an expanded mCA clone (1,620 autosomal). In the BBJ cohort, of 125,541 individuals without a history of hematologic cancer, only autosomal mCAs were available, with 20,440 carriers (16.3%), and 1,676 (1.3%) who carried an expanded clone. In the CUB COVID-19 cohort, of 871 individuals without a history of hematologic cancer, 258 (29.6%) carried an mCA (168 autosomal) and 177 (20.3%) carried an expanded mCA clone (128 autosomal) (Supplementary Table 2).
Consistent with previous reports, the prevalence of mCAs increased with age and they were more common in men (Supplementary Figs. 8 and 9 and Supplementary Table 3). Across the UKB, MGBB, FinnGen and BBJ cohorts combined, the prevalence of expanded mCAs was 0.5% in individuals aged <40 years, 1.2% in individuals aged 40–60 years, 7.8% in individuals aged 60–80 years, and 26.5% in those aged >80 years (Fig. 1b), the majority of which is due to the loss of the X chromosome (chrX) in female individuals and the loss of the Y chromosome (chrY) in male individuals (Supplementary Fig. 8). The prevalence of expanded autosomal mCAs was 0.27% for individuals aged <40 years, 0.52% for those aged 40–60 years, 1.5% for those aged 60–80 years, and 4.6% for those aged >80 years (Fig. 1c).
Association of mCAs with hematologic traits
We observed a striking association of mCA cell fraction with aberrant cell blood counts in blood samples acquired at the same visit as blood for genotyping (Fig. 2a,b). Increased mCA cell fraction was associated with overall increased white blood cell count, with general consistency across the cell differential components, and inflections at around a cell fraction of 0.1 (Fig. 2b). The strongest association across all mCA groupings (autosomal, chrX, chrY) with blood counts was the association between expanded autosomal mCAs and increased lymphocyte count at enrollment (β = 0.40 s.d. or 0.25 × 109 cells l−1; 95% confidence interval (CI) = 0.36–0.44 s.d.; P = 4.2 × 10−84) (Fig. 2a and Supplementary Fig. 10).

a, Linear regression of the association between blood counts and expanded mCAs. Associations are adjusted for age, age2, sex, smoking status and principal components of ancestry. Error bars show the 95% CI for estimates, and the Bonferroni correction was used to determine the level of statistical significance. b, The relationship of mCA cell fraction with blood counts (in units of 109 cells l−1) in the UKB in individuals without prevalent hematologic cancer at the time of blood draw for genotyping and cell count measurement. The dotted horizontal lines reflect the mean blood count for individuals without an mCA. The dotted vertical lines at the cell fraction of 0.10 represent the cut-off for the definition of expanded mCA. Individuals with known hematologic cancer at the time of or prior to blood draw for genotyping were excluded. Error bands reflect the standard error of a generalized additive model with integrated smoothness fit to the data. c, Association of expanded mCA categories (with cell fraction > 10%) with incident cancer in the UKB. Analyses are adjusted for age, age2, sex, smoking status and principal components of ancestry. Individuals with a history of hematologic cancer at enrollment were removed from the analysis. Error bands are derived from binomial proportion standard errors. d, Assessment of the association of the expanded mCA categories (with cell fraction > 10%) with incident cancer in the UKB, using Cox proportional hazards modeling, with time-on-study as the underlying timescale. Analyses are adjusted for age, age2, sex, smoking status and principal components of ancestry. Error bars show the 95% CI for estimates, and the Bonferroni correction was used to determine the level of statistical significance. Individuals with a history of hematologic cancer at enrollment were removed from analysis. AML, acute myeloid leukemia; MPN, myeloproliferative neoplasm; RBC, red blood cell; WBC, white blood cell.
Similarly, incident hematologic cancer risk was also strongly dependent on cell fraction (Fig. 2c). We reproduced the associations of mCAs with hematologic cancers that had previously been identified in the UKB data11,12. We found that expanded autosomal mCAs with cell fraction >10% were most strongly associated with incident hematologic cancer (Fig. 2d), with the strongest association being for incident chronic lymphocytic leukemia (CLL) (hazard ratio (HR) 120.48; 95% CI = 92.53–156.86; P = 2.2 × 10−277), although an association with polycythemia vera (HR 32.56; 95% CI = 22.81–46.48; P = 6.0 × 10−82) and with myeloid leukemia (HR 11.82; 95% CI = 7.29–19.18; P = 1.4 × 10−23) was also present (Fig. 2d). In comparison, the associations of chrX and chrY mCAs with CLL were considerably weaker (chrX: HR 27.40; 95% CI = 6.58–114.16; P = 5.5 × 10−6; chrY: HR 1.91; 95% CI = 0.96–3.80; P = 0.064) (Fig. 2d).
Associations with diverse infections
Across the genome, the presence of any mCA was associated with diverse incident infections (defined in Supplementary Tables 4 and 5) (HR 1.06; 95% CI = 1.04–1.09; P = 8.6 × 10−8) (Supplementary Fig. 11), independent of age, age2, sex, smoking status and the first ten principal components of ancestry in the combined UKB, MGBB and FinnGen meta-analysis. The dependence of this association on mCA cell fraction is further demonstrated in Fig. 3a,b, which shows an increase in the proportion of incident infection cases and incident sepsis cases with cell fraction, with greater slopes observed at a cell fraction of >10%. Accordingly, the associations across diverse infections were stronger for expanded mCA clones (HR 1.12; 95% CI = 1.07–1.17; P = 6.3 × 10−7) (Fig. 3c). Furthermore, of the expanded mCA clones, the strongest association was observed for expanded autosomal mCAs (HR 1.25; 95% CI = 1.15–1.36; P = 1.8 × 10−7) (Fig. 3c). To account for multiple hypothesis-testing, expanded autosomal mCAs were significantly associated with sepsis (HR 2.68; 95% CI = 2.25–3.19; P = 3.1 × 10−28), respiratory system infections (HR 1.36; 95% CI = 1.24–1.50; P = 3.8 × 10−10), digestive system infections (HR 1.51; 95% CI = 1.32–1.73; P = 2.2 × 10−9) and genitourinary system infections (HR 1.25; 95% CI = 1.11–1.41; P = 3.7 × 10−4) (Fig. 3c). The specific expanded autosomal mCAs implicated for infection were diverse in nature (across all chromosomes, of different sizes), and included a mix of copy number gain, loss, and CNN-LOH mutations (Extended Data Fig. 2). Further associations across 20 specific infectious disease subcategories are enumerated in Extended Data Fig. 3. For sex chromosome mCAs, none of the incident infections achieved statistical significance (that is, P < 0.005) in a meta-analysis across the three cohorts; however, there was a trend towards an association with respiratory infections (expanded chrX: HR 1.45; 95% CI = 1.11–1.90; P = 0.0068; expanded chrY: HR 1.09; 95% CI = 1.03–1.16; P = 0.005) (Extended Data Fig. 4).

a,b, The proportion of individuals in the UKB with any incident infection or sepsis, according to cell fraction, for all mCAs (a) and autosomal mCAs (b), in individuals without prevalent hematologic cancer at the time of blood draw for genotyping. The dotted vertical lines at a cell fraction of 0.10 represent the cut-off for the definition of expanded mCAs. Error bands are derived from binomial proportion standard errors. c, The association of any expanded mCA, and separately, expanded autosomal mCAs, with incident infections across individuals in the UKB, MGBB and FinnGen cohorts, using Cox proportional hazards modeling with the underlying timescale of time-on-study. Analyses are adjusted for age, age2, sex, smoking status and principal components 1–10 of ancestry. Error bars show the 95% CI for estimates, and the Bonferroni correction was used to determine the level of statistical significance. Individuals with prevalent hematologic cancer were excluded from analysis. Association analyses for other groupings of mCAs (including across all mCAs regardless of cell fraction, as well as chrX and chrY mCAs) are provided in Supplementary Figs. 11 and 12. BBJ, BioBank Japan; mCA, mosaic chromosomal alterations; MGBB, Mass General Brigham Biobank; UKB, UK Biobank.
Risks for incident fatal infections were assessed in the BBJ cohort given that non-fatal incident infectious disease events are currently unavailable for BBJ. For individuals without any cancer history in the BBJ cohort, autosomal mCAs had nominal associations with fatal incident infections (HR 1.12; 95% CI = 1.0–1.2; P = 0.04), with expanded autosomal mCAs being associated with incident sepsis mortality (HR 2.04; 95% CI = 1.04–4.16; P = 0.05) (Supplementary Table 6 and Extended Data Fig. 5), as well as with pneumonia history (odds ratio (OR) 1.40; 95% CI = 1.12–1.53; P = 0.00080).
In a sensitivity analysis of the association of expanded autosomal mCAs and incident sepsis, the association was consistently significant across different age groups (Supplementary Fig. 12), and it was additionally independent of a 25-factor smoking covariate17, body mass index, type 2 diabetes mellitus, leukocyte count, lymphocyte count and lymphocyte percentage (Supplementary Table 7).
Stratified analyses indicated that expanded autosomal mCAs in individuals with cancer prior to infection (either any solid tumors or hematologic malignancy after the time of blood draw for genotyping) conferred a stronger association with sepsis (HR 2.79; 95% CI = 2.30–3.38; P = 9.7 × 10−26) and respiratory system infections (HR 1.60; 95% CI = 1.40–1.82; P = 6.1 × 10−12) compared with individuals without a prior cancer history (sepsis: HR 1.25; 95% CI = 0.80–1.95; P = 0.33, Pinteraction = 0.001; respiratory system infections: HR 1.16; 95% CI = 1.00–1.34; P = 0.045, Pinteraction = 0.001) (Fig. 4 and Supplementary Figs. 13–15). This interaction was driven by prevalent solid cancer, not hematologic cancer, after DNA acquisition for mCA genotyping (Supplementary Table 8). Further multivariable adjustment indicated that incident sepsis and infection were independent of chemotherapy, neutropenia, aplastic anemia, decreased white blood cell count, bone marrow or stem cell transplant, and radiation effects prior to infection (with these phenotypes defined using International Classification of Diseases tenth revision (ICD-10) and ICD-9 phecode groupings19) (Supplementary Table 9). We also explored the time difference between cancer diagnosis and specific infections to characterize the potential influence of expanded mCA. Univariable analyses showed that expanded mCA carriers tend to have a twofold higher incidence of post-cancer diagnosis septicemia and pneumonia, and that the difference in incidence rate was more prominent in infections occurring >3 years after the cancer diagnosis (Supplementary Table 10 and Supplementary Fig. 16). Besides cancer patients, we also calculated the univariable association between expanded mCA and diseases in the general public. On average, if we followed individuals without documented cancer, sepsis, or pneumonia history in the UKB for 1,000 person-years after expanded mCA detection, we would observe that 36 individuals develop incident cancer (5 cases of which would be hematological cancer), 14 individuals develop incident pneumonia, and 8 develop incident sepsis (Extended Data Fig. 6).

a, The association of expanded autosomal mCAs with incident infections across individuals with and without a cancer history before their incident infection, in a meta-analysis of the UKB, MGBB and FinnGen cohorts combined (cohort-specific analyses are available in Supplementary Fig. 14), assuming a fixed effect. Error bars show the 95% CI for estimates, and the Bonferroni correction was used to determine the level of statistical significance. Individuals with known hematologic cancer at the time of or prior to blood draw for genotyping were excluded. Analyses are adjusted for age, age2, sex, smoking status and principal components of ancestry. b, Cumulative incidence curves for various infections in the UKB. (Results from the MGBB and FinnGen are available in Supplementary Fig. 16.) Red, mCA positive and cancer positive; purple, mCA negative and cancer positive; blue, mCA positive and cancer negative; green, mCA negative and cancer negative. Individuals with known hematologic cancer at the time of or prior to blood draw for genotyping were excluded.
Association with COVID-19 severity
Across 719 cases of hospitalization for COVID-19 in the UKB, 44 individuals (6%) carried an expanded mCA clone at the time of enrollment (in 2010), compared with 3% of 337,877 controls. Adjusting for age, age2, sex, prior or current smoking status and principal components of ancestry, expanded mCAs were associated with COVID-19 hospitalizations (OR 1.59; 95% CI = 1.13–2.25; P = 0.0082), with higher effect estimates conferred by expanded autosomal mCAs (OR 2.17; 95% CI = 1.16–4.08; P = 0.016) (Fig. 5a). Analyses in the FinnGen cohort showed evidence of independent replication. The meta-analyzed associations across the UKB and FinnGen of expanded autosomal mCAs on COVID-19 hospitalization were OR 2.44 (95% CI 1.33 to 4.46; P = 0.0038). In the UKB, further sensitivity analysis was performed; the associations persisted with additional adjustment for normalized Townsend deprivation index, normalized body mass index, type 2 diabetes mellitus, hypertension, coronary artery disease, and any cancer, asthma, and chronic obstructive pulmonary disease (Extended Data Fig. 7a). Additionally, similar associations were observed in the UKB when comparing COVID-19 hospitalization with tested negative controls, COVID-19 positive with all participants from English provinces, and COVID-19 positive with tested negative controls (Extended Data Fig. 7b). Similar to the diverse nature of mCA clones observed in cases of incident infection, specific mCA clones carried by individuals hospitalized with COVID-19 were also diverse in nature: across multiple chromosomes, a wide range of sizes, and across copy number gain, loss, and CNN-LOH (Fig. 5b). Similar associations of expanded mCAs with COVID-19 were also observed for incident pneumonia in the UKB (Extended Data Fig. 7c).

a, The association of expanded mCAs with COVID-19 hospitalization across the UKB and FinnGen cohorts, determined by logistic regression. Error bars show the 95% CI for estimates, and the Bonferroni correction was used to determine the level of statistical significance. Individuals with known hematologic cancer at the time of or prior to blood draw for genotyping were excluded. Analyses are adjusted for age, age2, sex, ever smoking status and the principal components of ancestry. b, Visual representation of the diverse range of expanded autosomal mCAs detected across the genome in individuals hospitalized with COVID-19 in the UKB cohort. Each point represents one mCA carried by a hospitalized individual with COVID-19. c. Proportion of expanded autosomal mCAs in each category of COVID-19 outcomes for the CUB COVID-19 cohort, defined using the WHO COVID-19 scale (n = 871 participants). The binomial proportion 95% CIs are shown. In the CUB cohort, the OR of the adjusted association between expanded autosomal mCAs and these ordinal COVID-19 outcomes, evaluated using ordinal regression, is 1.52 (95% CI = 1.04–2.21; P = 0.031, two-tailed). The summary statistics for the covariates included in the adjusted model for the CUB cohort are listed in Supplementary Table 11. CNN-LOH, copy number neutral loss of heterozygosity; CUB, Columbia University Biobank; MGBB, Mass General Brigham Biobank; UKB, UK Biobank; WHO, World Health Organization.
We next identified 871 patients with COVID-19 from the CUB and classified them into mutually exclusive ordinal categories based on COVID-19 outcomes and the World Health Organization (WHO) COVID-19 progression scales: mild cases (n = 52), COVID-19 infection not requiring hospitalization (WHO stages 1–3); moderate cases (n = 440), COVID-19 infection requiring hospitalization but without intubation or death (WHO stages 4–6); and severe cases (n = 379), respiratory failure due to COVID-19 requiring endotracheal intubation and mechanical ventilation (n = 140; WHO stages 7–9) or death from COVID-19 (n = 239; WHO stage 10). Individuals with prevalent hematologic cancer were excluded from analyses as before. Expanded autosomal mCAs were detected in 5.8% of patients with mild cases, in 13.9% of patients with moderate cases, and in 16.9% of patients with severe cases (Fig. 5c). Expanded autosomal mCAs were associated with these ordinal COVID-19 outcomes with an OR of 1.52 (95% CI = 1.04–2.21; P = 0.031), adjusted for age, sex and self-reported ancestry. Summary statistics for the multivariate logistic regression are listed in Supplementary Table 11. This association was also independent of the status of any other prevalent cancers, as validated by a sensitivity analysis that includes adjustment for any cancer diagnosis (Supplementary Table 12).
Germline genetic predisposition to expanded mCAs
To further elucidate causal factors for expanded mCA clones, we performed a GWAS in the UKB cohort. We identified 63 independent genome-wide significant loci (r2 < 0.1 across 1 megabase (Mb) windows of the genome) (Fig. 6a and Supplementary Table 13). Across the 63 germline variants, significant correlation was seen between different mCA categories (Extended Data Fig. 8), suggesting the presence of shared germline genetic variants predisposing to mCAs across the genome. Follow-up analyses using an additive polygenic risk score consisting of 156 independent genome-wide significant variants associated with mosaic loss of chromosome Y (mLOY) from male participants from a prior study in the UKB20, found significant associations with expanded autosomal mCAs and expanded chrX mCAs in female participants, further highlighting the shared germline contributors towards mCAs across the genome (Extended Data Fig. 9). The association of the 156 previously identified independent genome-wide significant variants associated with mLOY20 with the expanded chrY mCA categories in the UKB cohort shows that the two are highly correlated (rp = 0.91; P = 3.80 × 10−57), with 1.87-fold higher effect estimates conferred on expanded chrY mCAs compared with all mLOY variants20 (Supplementary Fig. 17). Additionally, a strong correlation is seen between germline variants associated with mLOY and their associations with expanded chrX mCAs, expanded autosomal mCAs, and all expanded mCAs (Supplementary Fig. 17). Further analysis of the TP53 variant rs78378222-G identified a particularly strong effect on expanded chrY mCAs (OR 2.03; 95% CI = 1.79–2.31; P = 1.33 × 10−27) in addition to all chrY mCAs (OR 1.79; 95% CI = 1.66–1.92; P = 8.81 × 10−53), with the chrY mCA effect being very similar to that previously reported by Thompson et al.20 (Supplementary Table 14). The TP53 variant rs78378222-G was also associated with expanded autosomal mCAs (OR 1.51; 95% CI = 1.21–1.88; P = 0.00031) and expanded chrX mCAs (OR 2.26; 95% CI = 1.30–3.92; P = 0.0038). The autosomal mCAs carried by individuals with rs78378222-G were diverse in size, copy change and location in the genome (Supplementary Fig. 18). A transcriptome-wide association study (TWAS) combining the expanded mCA GWAS results with Genotype-Tissue Expression project version 8 (GTExv8; ref. 21) whole-blood expression quantitative trait loci using UTMOST (unified test for molecular signatures)22 prioritized 62 genes (P < 3.2 × 10−6) promoting expanded mCA development (Fig. 6b and Supplementary Table 15). Although gene enrichment analyses with the Elsevier Pathway Collection did not identify significantly associated pathways after multiple testing correction, the top pathways were linked to DNA damage repair and lymphoid processes (Extended Data Fig. 10a, Supplementary Table 16). The corresponding GWAS LocusZoom plots for some of these immune-related genes are shown in Extended Data Fig. 10b. To prioritize the tissues that were most strongly implicated by these loci, tissue enrichment analyses using GenoSkyline-Plus were performed. Significant enrichment was identified in immune-specific epigenetic and transcriptomic functional regions of the genome (P = 7.1 × 10−9) (Fig. 6c). Further stratification of the immune category identified specific enrichment for CD4+ T cells (P = 0.00098) (Fig. 6d).

a, GWAS for expanded mCA identified 63 independent loci. b, Quantile–quantile plot of the whole-blood TWAS of the expanded mCA GWAS using 670 samples from GTExv8, showing significant enrichment across 62 genes. The horizontal dotted line reflects the Bonferroni-adjusted P value for significance. Genes with P < 5 × 10−8 in the TWAS or those important in the pathway enrichment analyses from Extended Data Fig. 10 are labeled. c, Cell-type enrichment results from the expanded mCA GWAS across immune, brain, cardiovascular, muscle, gastrointestinal, epithelium, and other tissues, as annotated using GenoSkyline-Plus annotations. d, A zoom-in of c to show the stratified enrichment by specific categories of immune cells and tissues. In c and d, the vertical dashed lines indicate P = 0.05 for suggestive enrichment, and the Bonferroni-adjusted P-value for significant enrichment.
Discussion
Across five geographically distinct biobanks of data on 768,762 individuals without known hematologic malignancy, clonal hematopoiesis, represented by expanded mCAs, is increasingly prevalent with age but is not readily detectable by conventional medical blood tests. In addition to strongly predicting the future risk of hematologic malignancy, expanded mCAs are also associated with the risk for diverse incident infections, particularly sepsis and respiratory infections. These findings are robust across age, sex and tobacco smoking, and are strongest for those who develop cancer. Consistent with these observations, expanded mCAs are also associated with increased odds for COVID-19 hospitalization.
These results support several conclusions. First, mCA-driven clonal hematopoiesis is a potential risk factor for infection. Recent work has shown that clonal hematopoiesis with myeloid malignancy driver mutations, also referred to as ‘clonal hematopoiesis of indeterminate potential’, predisposes to myeloid malignancy and coronary artery disease23,24,25,26,27. Meanwhile, clonal hematopoiesis with larger chromosomal alterations (that is, mCAs) predisposes primarily to lymphoid malignancy but not coronary artery disease10,11,12,15,16. Our observations suggest that clonal hematopoiesis (defined as the presence of mCAs) is a risk factor for infection. Given that the relationship between mCAs and infection risk was not substantially attenuated when adjusting for leukocyte or lymphocyte counts at the baseline visit, the impact of mCAs on infection risk possibly acts through mechanisms independent of the impact of clonal hematopoiesis on cell counts. As an example, given that mCAs alter gene dosage (for example, via duplications and deletions) and remove allelic heterogeneity (for example, CNN-LOH events) in leukocytes, potential impacts on the differentiation, function and survival of leukocytes are mechanisms that could lead to altered infection risk. Our germline analyses specifically implicate lymphoid tissues. In particular, many of the mCA susceptibility loci are the same as those found in CLL, a condition in which lymphocyte differentiation and function are altered, promoting infection risk28,29,30,31. Therefore, molecular changes in leukocytes that promote clonal expansion may occur at the expense of reduced ability to combat infection.
Second, the infectious disease risk associated with mCAs is exacerbated in the setting of cancer. It is well-established that mCAs in blood-derived DNA increase the risk for hematologic cancer10,11,12. Furthermore, recent evidence suggests an association between mCAs detected in blood-derived DNA and an increased risk of select solid tumor14,17,32. Our analysis identified an interaction between mCAs and prior cancer diagnosis that amplified the sepsis and pneumonia risk. Importantly, this interaction was restricted to individuals with solid cancers, not antecedent blood cancer. Although this observation could be partially due to synergistic immunosuppressive side-effects of cancer therapies33, the observed associations persisted despite adjustment for many of these treatments. Alternatively, abnormal regulation of immune inflammatory pathways that release cytokines and inflammatory cells may create chronic states of inflammation in individuals with mCAs34,35. Based on our analysis, carriers of autosomal mCA are at an increased risk for sepsis (2.7-fold), pneumonia (1.8-fold), respiratory system infections (1.4-fold), digestive system infections (1.5-fold) and genitourinary system infections (1.3-fold), and these effects are more prominent in cancer patients. Surveillance for expanded mCA clones, particularly for those who develop solid cancer, may help identify individuals at high risk for infection who could benefit from targeted interventions.
Third, our findings could have particular relevance for the ongoing COVID-19 pandemic. We observed that mCAs are associated with elevated risk for COVID-19 hospitalization, with a greater than twofold risk linked to expanded autosomal mCAs. Maladaptive immune responses, particularly in leukocytes, increase the risk for severe COVID-19 infections36,37,38,39. Awareness of the COVID-19 risk associated with mCAs may help with the prioritization of prophylactic treatments. However, the question of whether immune response to current vaccination approaches is altered in the context of mCAs deserves further study.
Last, the mCA germline genetic associations identified in the present study replicate many of those previously identified10,11,20 and additionally suggest a common heritable basis across mCA classes, which may inform therapeutic targets. Genetic variants that influence the risk of autosomal mCAs also influence the risk of chrX mCAs in female individuals and that of chrY mCAs in male individuals. Furthermore, previously published genetic variants associated with mLOY20 also influence the risk of autosomal mCAs and chrX mCAs in female individuals. These loci may support putative therapeutic targets that may decrease the risk of mCA development, the rate of mCA clonal expansion, or the risk of progression of mCAs to clinical outcomes.
This analysis of mCAs and infection had some limitations. First, our study measures mCAs only at one time point for each participant. Although our sampled mCA time point is probably correlated with clonal hematopoiesis at the time of infection, clonal hematopoiesis dynamically changes over time, potentially leading to differences in cellular fraction or additional undetected events that were acquired prior to infection. Second, we cannot rule out the possibility of undiagnosed hematologic malignancy in individuals with mCAs with only blood DNA. However, given the observed prevalence of mCAs (4% by age 60 years) in individuals without diagnosed hematologic malignancy and the general scarcity of hematologic malignancy in the general population, we anticipate that undiagnosed hematologic malignancy at DNA acquisition will be uncommon. Third, despite the robust adjustment and sensitivity analyses performed in the statistical analysis, including adjustment for cancer subtype, chemotherapy, bone marrow transplant, radiation, and other features associated with poor cancer prognosis (neutropenia, aplastic anemia, decreased white blood cell count), we cannot completely rule out the impact of residual confounding from unknown or unmeasured sources on the results. Here, consistency across cohorts and infection types, and biologic plausibility mitigate this possibility, and the empiric association of mCAs with incident infection may enable improved clinical risk prediction in patient populations as further scientific work is performed to understand the biological mechanisms by which mCAs influence the immune system. Last, further causal inference analyses using methods such as Mendelian randomization are limited by the low heritability of autosomal mCAs11 and the low heritability of infectious diseases40,41. However, defects in humoral, cell-mediated and innate immunity have been linked to CLL28,29,30,31. Whether all of these or specific aspects of immunity are altered for this pre-CLL condition requires further study.
In conclusion, we report evidence for increased susceptibility to a spectrum of infectious diseases in individuals carrying autosomal mCAs in a detectable fraction of leukocytes. The impacts of mCA on infection risk are systemic, with increased susceptibility to infection observed for a variety of organ systems, including severe COVID-19 presentations.
Methods
Study samples
The UKB, a population-based cohort of approximately 500,000 participants recruited from 2006 to 2010, has existing genomic and longitudinal phenotypic data42. Baseline assessments were conducted at 22 assessment centers across the United Kingdom, with sample collections including blood-derived DNA. Of 488,377 genotyped individuals, we analyzed 445,101 participants who consented to genetic analyses and who passed sample quality control criteria for mCA calling, had genotypic–phenotypic sex concordance, no first- or second-degree relatives (random exclusion of one from each pair), and no prevalent hematologic cancer at the time of blood draw. Genome-wide genotyping of blood-derived DNA was performed by UKB using two genotyping arrays sharing 95% of marker content: Applied Biosystems UK BiLEVE Axiom Array (807,411 markers in 49,950 participants) and Applied Biosystems UK Biobank Axiom Array (825,927 markers in 438,427 participants), both by Affymetrix42. Secondary use of the data was approved by the Massachusetts General Hospital Institutional Review Board (protocol 2013P001840) and was facilitated through UKB applications 7089 and 21552.
The MGBB contains genotypic and clinical data from >105,000 patients who consented to broad-based research across seven regional hospitals43. Baseline phenotypes were ascertained from the electronic medical record and from surveys on lifestyle, environment, and family history. Of the approximately 36,000 genotyped individuals, 27,778 samples had available probe raw intensity data (IDAT) files for mCA calling. Blood-derived DNA samples were genotyped using three versions of the Multi-Ethnic Genotyping Array (MEGA) Single-Nucleotide Polymorphism (SNP) array offered by Illumina. Secondary use of the data was approved by the Massachusetts General Hospital Institutional Review Board (protocol 2020P000904).
The FinnGen project (https://www.finngen.fi/en), launched in 2017, covers the whole of Finland and aims to improve the health of people around the world through genetic studies. The latest released version (R6) contains genotypic, demographic and extensive health information (for example, a national inpatient register since 1969 and a national outpatient register since 1998, a cancer register since 1953, and a drug reimbursement register since 1964) from 269,077 Finnish individuals. Blood-derived DNA samples were genotyped using two versions of FinnGen Thermo Fisher Axiom custom array (https://www.finngen.fi/en/researchers/genotyping) provided by the Thermo Fisher genotyping service facility.
The BBJ is a hospital-based registry that collected clinical, DNA, and serum samples from approximately 200,000 consenting patients with one or more of 47 target diseases at a total of 66 hospitals between 2003 and 2007 (ref. 44). Blood DNA was genotyped in three batches using different arrays or a set of arrays, namely: (1) a combination of Illumina Infinium Omni Express and Human Exome; (2) Infinium Omni Express Exome v.1.0; and (3) Infinium Omni Express Exome v.1.2, which capture very similar SNPs. These analyses were approved by the ethics committees of RIKEN Center for Integrative Medical Sciences and the Institute of Medical Sciences, The University of Tokyo.
The CUB COVID-19 cohort includes multi-ethnic patients with COVID-19 who were treated at the Columbia University Irving Medical Center (CUIMC) and who underwent SNP genotyping on the Illumina Infinium Global Diversity Array. All patients in the cohort had a polymerase chain reaction-confirmed SARS-CoV-2 infection. All patients who had a blood draw at CUIMC after their positive polymerase chain reaction test were recruited regardless of hospitalization status. These patients were recruited to the CUB between March and May 2020, at the peak of the first wave of the New York City pandemic, thus only a small fraction of the cohort was not hospitalized. The CUB COVID-19 studies are reviewed and approved by the Columbia University Medical Center Institutional Review Board. A subset of patients was included under a public health crisis institutional review board waiver of consent specifically for COVID-19 studies if patients were deceased, not able to consent, or if the study team was unable to contact them as per the Columbia Institutional Review Board protocols AAAS9552 and AAAS7370. The primary analysis involved 871 patients and excluded individuals who had hematological malignancies. This cohort (n = 871) was composed of 480 male participants and 391 female participants; the average age was 62 years (range, 7–101 years); 52% of the participants self-reported as being Hispanic or Latinx, 14% self-reported as being Black or African American, 11% self-reported as being white or European and 23% self-reported as other or unknown. All COVID-19-positive patients were classified into mutually exclusive ordinal outcome categories as defined by the WHO: mild cases (n = 52), COVID-19 infection not requiring hospitalization (WHO stages 1–3); moderate cases (n = 440), COVID-19 infection requiring hospitalization but without intubation or death (WHO stages 4–6); and severe cases (n = 379), respiratory failure due to COVID-19 requiring endotracheal intubation and mechanical ventilation (n = 140; WHO stages 7–9) or death from COVID-19 (n = 239; WHO stage 10).
Mosaic chromosomal alteration detection
The detection of mCAs in the UKB has been described previously11,12. In brief, genotype intensities were transformed to log2 R ratio (LRR) and B-allele frequency (BAF) values to estimate total and relative allelic intensities, respectively. Re-phasing was performed using Eagle2 (ref. 45), and mCA calling was performed by leveraging long-range phase information to search for allelic imbalances between maternal and paternal allelic fractions across contiguous genomic segments. Constitutional duplications and low-quality calls were filtered out and cell fraction was estimated as previously described12. UKB mCA calls were obtained from dataset Return 2062 generated from UKB application 19808.
Detection of mCAs in the MGBB was performed starting from raw IDAT files from the Illumina MEGA. Genotype clustering was performed using the Illumina GenCall algorithm. The resulting GTC genotype files were converted to VCF files using the bcftools gtc2vcf plugin (https://github.com/freeseek/gtc2vcf). Genotype phasing across the whole cohort was performed using SHAPEIT4 (ref. 4) in windows of a maximum of 20 cM, with an overlap of 2 cM between consecutive windows. Phased genotypes were ligated across overlapping windows using bcftools concat (https://github.com/samtools/bcftools). mCA detection in the MGBB was performed using MoChA1,2 (https://github.com/freeseek/mocha). A pipeline to execute the whole workflow from raw files all the way to final mCA calls is available in WDL (workflow description language) format for the Cromwell execution engine46 as part of MoChA. We excluded 160 samples with phased BAF auto-correlation >0.05, which is indicative of contamination or of other potential sources of poor DNA quality, and 67 samples with phenotype–genotype sex discordance (Supplementary Fig. 1). We removed probable germline copy number polymorphisms (lod_baf_phase <20 for autosomal variants and lod_baf_phase <5 for sex chromosome variants), constitutional or inborn duplications (mCAs of <2 Mb with relative coverage >2.25, and mCAs of 2–10 Mb with relative coverage >2.4) and deletions (filtering out mCAs with relative coverage <0.5) (Supplementary Fig. 2).
FinnGen blood samples are genotyped using two versions of the FinnGen Thermo Fisher Axiom custom array. The detection of mCAs in FinnGen was performed, starting from the genotype–intensity tables of 201,322 samples using the ‘txt’ mode of the MoChA WDL pipeline (https://github.com/freeseek/mocha). The input genotype–intensity tables for mCA detection were directly provided by the Thermo Fisher genotyping service, which performed genotype calling from the raw CEL files for each batch using the apt-probeset-genotype tool. Genotype phasing across the whole cohort was performed using SHAPEIT4 in windows of a maximum of 20 cM, with 2 cM of overlap between consecutive windows. Phased genotypes were ligated across overlapping windows using bcftools concat (https://github.com/samtools/bcftools). We excluded 215 samples with phased BAF auto-correlation >0.05, which is indicative of contamination or of other potential sources of poor DNA quality, and 83 samples with phenotype–genotype sex discordance (Supplementary Fig. 3). We removed probable germline copy number polymorphisms (lod_baf_phase <20 for autosomal variants and lod_baf_phase <5 for sex chromosome variants, and lod_baf_phase <10 unless they are larger than 5 Mb (or 10 Mb if they span the centromere)), constitutional or inborn duplications (0.5–5 Mb mCAs with relative coverage >2.5 and Bdev < 0.1, and 5–10 Mb mCAs with relative coverage >2.75) and deletions (filtering out mCAs with relative coverage <0.5) (Supplementary Fig. 4). After further removing first- or second-degree relatives, and individuals with any prevalent hematologic cancer history at the time of blood draw for genotyping, there were 175,690 samples remaining for analyses.
The detection of mCAs in the BBJ has been described previously10. In brief, genotyping intensity data were analyzed across variants shared between the three primary arrays, and were used to compute BAF and LRR values. Phasing was performed using the Eagle2 software. Mosaic events were called as previously described12.
The CUB COVID-19 blood samples were genotyped using the Illumina Infinium Global Diversity Array. Detection of mCAs was performed starting from the probe raw IDAT files of 1,182 samples. The resulting raw intensity data were converted to VCF files using the bcftools gtc2vcf plugin (https://github.com/freeseek/gtc2vcf). Genotype phasing was performed using Eagle2 over the entire cohort. After excluding samples with a call rate of <0.97 and further removing first- or second-degree relatives, the mCA calling was performed using the MoChA pipeline (https://github.com/freeseek/mocha). We excluded 133 samples with phased BAF auto-correlation >0.05, indicative of contamination or of other potential sources of poor DNA quality, and six samples with phenotype–genotype sex discordance (Supplementary Fig. 5). We removed probable germline copy number polymorphisms (lod_baf_phase <20 for autosomal variants and lod_baf_phase <5 for sex chromosome variants), constitutional or inborn duplications (0–10 Mb mCAs with relative coverage >2.4) and deletions (filtering out mCAs with relative coverage <0.5) (Supplementary Fig. 6). We further excluded 32 individuals with any prevalent hematologic cancer history at the time of blood draw for genotyping and had 871 samples remaining for analyses.
Clinical outcomes
Definitions of infection outcomes are detailed in Supplementary Tables 4 and 5. In the UKB, the first reported occurrences over a median 8 year follow-up in category 2410 were used as categorized by the UKB, which maps primary care data, ICD-9 and ICD-10 codes from hospital inpatient data, ICD-10 codes in death register records, and self-reported medical conditions reported at the baseline, to ICD-10 codes. For each set of phenotypes grouped by organ system or by category, the time to first incident event after baseline examination in individuals free of prevalent history of each disease category was used. In the MGBB, electronic health record data were used to define incident ICD-10 codes grouped in the same fashion after DNA collection date over a median 3 year follow-up. In the FinnGen cohort, phenotypes were grouped together across ICD-8, ICD-9 and ICD-10 codes (Supplementary Table 2), with incident infections defined after DNA collection date over a median 3 year follow-up. In BBJ, analyses were performed using fatal incident events attributed to diverse infection outcomes in Supplementary Table 1, given that non-fatal incident events were not available. Additionally, analyses for pneumonia were performed using a history of pneumonia prior to genotyping, based on interviews and medical record reviews44. Cancer cases in the UKB were identified using the cancer register (category 100092) in combination with the inpatient ICD-10 registry (field identification numbers 41270 and 41280). Hematologic cancer cases in the UKB were identified using the cancer registry’s field identification number 40011 (hematological cancer identified from biopsy), the field identification numbers 40005 and 40006 in combination with the ICD-10 code ranges C81–96 and D45–47, and the inpatient ICD-10 registry (field identification numbers 41270 and 41280, in combination with the ICD-10 code ranges C81–96 and D45–47). In the MGBB, cancer cases were identified using the ICD-10 code range C00–D49, and hematologic cancer cases were identified using the ICD-10 code ranges C81–96 and D45–47. Other clinical phenotypes defined in the UKB, MGBB, and FinnGen cohort are detailed in Supplementary Tables 17–19. Smoking status in the MGBB was defined using a combination of electronic health record data and survey data. Follow-up time was coded as the time from blood draw for genotyping to the event (development of incident phenotype) or, for controls, as the time from sample collection to either the censor date (31 October 2019) or the date of death if the patient died prior to the last censor. Smoking status in the FinnGen cohort was defined based on survey data. Follow-up time was coded as the time from blood draw for genotyping to the event (development of incident phenotype) or, for controls, as the time from sample collection to either the censor date (31 December 2019) or the date of death if the patient died prior to the last censor.
UKB COVID-19, from SARS-CoV-2 infection, phenotypes used in the present analysis were downloaded on 27 July 2020. SARS-CoV-2 infection was determined using polymerase chain reaction testing of nasopharyngeal, oropharyngeal or lower respiratory samples obtained between 16 March 2020 and 17 July 2020. Patients with COVID-19 requiring hospitalization were defined as any individual with at least one positive test who also had evidence for inpatient hospitalization (field identification number 40100). The controls included two sets: participants from UKB English recruitment centers who were not known to have COVID-19 (that is, individuals with negative or no known SARS-CoV-2 testing); or participants with a negative SARS-CoV-2 test. Individuals with COVID-19 of unknown or low severity (that is, individuals who had at least one positive SARS-CoV-2 test without a known hospitalization) were excluded from the primary analyses.
Replication was performed in the FinnGen cohort when SARS-CoV-2 infection was confirmed, either by polymerase chain reaction testing or by identification of antibodies in samples obtained between 2 March 2020 and 27 July 2020. Across both the UKB and FinnGen cohorts, individuals who died prior to 1 March 2020, and therefore were not at risk for COVID-19 infection, were excluded from COVID-19 analyses.
Statistical methods for infection associations
Analyses of the association of expanded mCAs with primary incident infection across the ten main infectious disease organ system categories (listed under ‘organ system’ in Supplementary Table 1) were performed using Cox proportional hazards models, adjusting for age, age2, sex, ever smoking status and principal components 1–10 from the genotyping data. The age2 term was added to account for potential quadratic associations between age and disease occurrence, given that the association between mCAs and age is also nonlinear. Time since DNA collection was used as the underlying timescale. The proportional hazards assumption was assessed using Schoenfeld residuals and was not rejected. Individuals with a history of hematological cancer prior to DNA collection were excluded. The threshold of significance used in the analyses of primary organ system infection was a two-sided Bonferroni threshold, P < 0.05/10 = 0.005, to account for multiple hypothesis-testing. Analyses of incident events were performed separately in each biobank using the survival package in R (version 3.5, R Foundation). Meta-analyses of the UKB, MGBB and FinnGen results were performed using a fixed-effects model from the meta package.
For the UKB COVID-19 analyses, logistic regression was performed to estimate the association between expanded mCAs and COVID-19 hospitalization using the aforementioned phenotype definition, adjusting for sex, age, age2, smoking status and the first ten principal components from the genotyping data. As above, individuals with prevalent hematologic cancer were excluded from analyses. For the COVID-19 analyses, statistical significance was assigned using a two-sided P value of <0.05. Secondary multivariable models were additionally adjusted for normalized Townsend deprivation index47, normalized body mass index at enrollment visit, and any prevalent or incident type 2 diabetes mellitus, hypertension, coronary artery disease, cancer, asthma and chronic obstructive pulmonary disease.
Further sensitivity analyses were performed to assess the associations between expanded autosomal mCAs and infection in the UKB. First, the associations of 20 incident infections with mCAs, across the ten broader organ system groups, were assessed using a Bonferroni threshold of significance (P < 0.05/20 = 0.0025). Second, stratified cancer analyses were performed in individuals with antecedent cancer prior to their incident infection in both the UKB and MGBB, additionally stratifying for the same aforementioned covariates (age, age2, sex, ever smoking status and the first ten principal components of genetic ancestry). Third, an interaction analysis was performed using an mCA × antecedent cancer term in the model to analyze the interaction between mCAs and antecedent cancer prior to incident infection. Fourth, for the incident sepsis association, four sets of covariates were added to the Cox proportional hazards model: (1) normalized body mass index and type 2 diabetes mellitus, (2) any antecedent cancer prior to incident infection, (3) adjustment for a more comprehensive 25-factor smoking phenotype17, and (4) adjustment for normalized leukocyte count, lymphocyte count and lymphocyte percentage at baseline visit. Fifth, we evaluated the association of expanded autosomal mCAs with incident sepsis and pneumonia in subgroups of individuals with cancer prior to infection (that is, those with prevalent solid cancer, incident hematologic cancer, and incident solid cancer prior to infection), in models adjusted for age, age2, sex, ever smoking status and the first ten principal components of genetic ancestry. Last, we further evaluated the association of expanded autosomal mCAs with incident pneumonia and sepsis in separate models adjusted for different predictors of cancer morbidity including chemotherapy, neutropenia, aplastic anemia, decreased white blood cell count, bone marrow or stem cell transplant, and radiation effects prior to infection (with these phenotypes defined using the Vanderbilt ICD-10 and ICD-9 phecode groupings19), in the same aforementioned models adjusted for age, age2, sex, ever smoking status and the first ten principal components of genetic ancestry.
Genome-wide association study
A GWAS was performed using Hail-0.2 (https://hail.is/) in the Google cloud. Variants were filtered to high-quality imputed variants (INFO score >0.4), with minor allele frequency >0.005, under Hardy–Weinberg equilibrium (P ≥ 1 × 10−10), as previously performed. A Wald logistic regression model was used for analysis, adjusting for age, age2, sex, ever smoking, principal components 1–10 and genotyping array. Significant, independent loci were identified using a threshold of significance of P < 5 × 10−8 and clumping in Plink-2.0, with an r2 threshold of 0.1 across 1 Mb genomic windows, using the 1000-Genomes Project European reference panel. An additive mLOY polygenic risk score was developed, \(\mathop {\sum}\nolimits_{i = 1}^{63} {{\mathrm{Beta}}\times {\mathrm{SNP}}_{ij}}\), where Beta is the weighting for each of the 156 independent genome-wide significant variants previously identified in the UKB male participants20, and SNPij is the number of alleles (that is, 0, 1 or 2) for SNPi in the female participant j in the UKB.
Cell-type enrichment analyses
We applied partitioned linkage disequilibrium (LD) score regression using LDSC48 (v1.0.1) to perform enrichment analysis using the expanded mCA GWAS summary statistics, in combination with tissue-specific epigenetic and transcriptomic functionality annotations from GenoSkyline-Plus22. In addition to the baseline annotations for diverse genomic features as suggested in the LDSC user manual, we specifically examined the enrichment signals in two tiers of annotations of different resolutions: GenoSkyline-Plus functionality scores of seven broad tissue clusters (immune, brain, cardiovascular, muscle, gastrointestinal tract, epithelial, and others); and GenoSkyline-Plus functionality scores of 11 tissue and cell types in the immune cluster (listed in Fig. 6d).
Transcriptome-wide association and pathway enrichment analysis
A TWAS was carried out using the expanded mCA GWAS summary statistics in combination with the UTMOST49 whole-blood model updated to GTExv8 (n = 670). Significant genes were identified using a Bonferroni cut-off of P < 0.05/15,625, or 3.2 × 10−6. Pathway enrichment analysis was performed using genes that had P < 0.001 in the TWAS, using the Elsevier Pathways through the EnrichR web server50.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
UKB individual-level data are available by request via application (https://www.ukbiobank.ac.uk). The mCA call set was previously returned to the UKB (return 2062) to enable individual-level linkage to approved UKB applications. Individual-level MGBB data are available from https://personalizedmedicine.partners.org/Biobank/Default.aspx, but restrictions apply to the availability of these data, which were used under institutional review board (IRB) approval for the current study, and so are not publicly available. The BBJ genotype data are available from the Japanese Genotype-phenotype Archive (JGA; http://trace.ddbj.nig.ac.jp/jga/index_e.html) under accession code JGAD00000000123. Individual-level linkage of mosaic events can be provided by the BBJ project upon request (https://biobankjp.org/english/index.html). FinnGen data may be accessed through Finnish Biobanks’ FinnBB portal (www.finbb.fi). Individual-level CUB COVID-19 data, including the mCA call set, are available by application from https://www.ps.columbia.edu/research/core-and-shared-facilities/core-facilities-category/columbia-university-biobank, but consent-related restrictions apply to the availability of these data, and data access requires separate IRB approval for the proposed data use. Aggregate data are also available upon reasonable request. Additionally, the full expanded mCA genome-wide association summary statistics have been uploaded onto the LocusZoom website (https://my.locuszoom.org/gwas/525823/). The present article includes all other data generated or analyzed during this study.
Code availability
A standalone software implementation (MoChA) of the algorithm used to call mCAs is available at https://github.com/freeseek/mocha. A pipeline to execute the whole workflow from raw files all the way to final mCA calls is available in WDL format for the Cromwell execution engine as part of MoChA. Code for all other computations is available upon request from the corresponding authors.
References
Gardner, I. D. The effect of aging on susceptibility to infection. Rev. Infect. Dis. 2, 801–810 (1980).
Gavazzi, G. & Krause, K. H. Ageing and infection. Lancet Infect. Dis. 2, 659–666 (2002).
Aw, D., Silva, A. B. & Palmer, D. B. Immunosenescence: emerging challenges for an ageing population. Immunology 120, 435–446 (2007).
Franceschi, C., Bonafe, M. & Valensin, S. Human immunosenescence: the prevailing of innate immunity, the failing of clonotypic immunity, and the filling of immunological space. Vaccine 18, 1717–1720 (2000).
Ongradi, J. & Kovesdi, V. Factors that may impact on immunosenescence: an appraisal. Immun. Ageing 7, 7 (2010).
Panda, A. et al. Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol. 30, 325–333 (2009).
Aoshi, T., Koyama, S., Kobiyama, K., Akira, S. & Ishii, K. J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 1, 226–232 (2011).
Holly, M. K., Diaz, K. & Smith, J. G. Defensins in viral infection and pathogenesis. Annu. Rev. Virol. 4, 369–391 (2017).
Pallett, L. J., Schmidt, N. & Schurich, A. T cell metabolism in chronic viral infection. Clin. Exp. Immunol. 197, 143–152 (2019).
Terao, C. et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature 584, 130–135 (2020).
Loh, P.-R., Genovese, G. & McCarroll, S. A. Monogenic and polygenic inheritance become instruments for clonal selection. Nature 584, 136–141 (2020).
Loh, P. R. et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355 (2018).
Lin, S. H. et al. Mosaic chromosome Y loss is associated with alterations in blood cell counts in UK Biobank men. Sci. Rep. 10, 3655 (2020).
Forsberg, L. A. et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 46, 624–628 (2014).
Jacobs, K. B. et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 44, 651–658 (2012).
Laurie, C. C. et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 44, 642–650 (2012).
Loftfield, E. et al. Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci. Rep. 8, 12316 (2018).
Machiela, M. J. et al. Characterization of large structural genetic mosaicism in human autosomes. Am. J. Hum. Genet. 96, 487–497 (2015).
Wu, P. et al. Mapping ICD-10 and ICD-10-CM codes to phecodes: workflow development and initial evaluation. JMIR Med. Inform. 7, e14325 (2019).
Thompson, D. J. et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657 (2019).
Consortium, G. T. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Lu, Q. et al. Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLoS Genet. 13, e1006933 (2017).
Bick, A. G. et al. Genetic interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 (2020).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Wang, L. et al. Integrated single-cell genetic and transcriptional analysis suggests novel drivers of chronic lymphocytic leukemia. Genome Res. 27, 1300–1311 (2017).
de Weerdt, I. et al. Innate lymphoid cells are expanded and functionally altered in chronic lymphocytic leukemia. Haematologica 101, e461–e464 (2016).
Bartik, M. M., Welker, D. & Kay, N. E. Impairments in immune cell function in B cell chronic lymphocytic leukemia. Semin. Oncol. 25, 27–33 (1998).
Arruga, F. et al. Immune response dysfunction in chronic lymphocytic leukemia: dissecting molecular mechanisms and microenvironmental conditions. Int. J. Mol. Sci. 21, 1825 (2020).
Zhou, W. et al. Mosaic loss of chromosome Y is associated with common variation near TCL1A. Nat. Genet. 48, 563–568 (2016).
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L. & Kroemer, G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28, 690–714 (2015).
Balkwill, F. & Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 357, 539–545 (2001).
de Visser, K. E., Eichten, A. & Coussens, L. M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37 (2006).
Lucas, C. et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature 584, 463–469 (2020).
Giamarellos-Bourboulis, E. J. et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27, 992–1000 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Cunha, L. L., Perazzio, S. F., Azzi, J., Cravedi, P. & Riella, L. V. Remodeling of the immune response with aging: immunosenescence and its potential Impact on COVID-19 immune response. Front. Immunol. 11, 1748 (2020).
Zekavat, S. M. et al. Elevated blood pressure increases pneumonia risk: epidemiological association and Mendelian randomization in the UK Biobank. Med (NY) 2, 137–148 (2021).
Tian, C. et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat. Commun. 8, 599 (2017).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Smoller, J. W. et al. An eMERGE clinical center at Partners Personalized Medicine. J. Pers. Med. 6, 5 (2016).
Nagai, A. et al. Overview of the BioBank Japan project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Voss, K., Auwera, G. & Gentry, J. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000Research https://doi.org/10.7490/f1000research.1114631.1 (2017).
Townsend, P., Phillimore, P. & Beattie, A. Health and Deprivation. Inequality and the North (Croom Helm, 1987).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Hu, Y. et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat. Genet. 51, 568–576 (2019).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Acknowledgements
The authors thank C. Whelan, C. Llanwarne, J. Cerrato, K. Vernest and K. Shakir, and many other members of the Terra/Cromwell team, for their help and advice in the development of the MoChA pipeline. The authors also thank P. Danecek for implementing critical features needed in BCFtools, S. Chanock for critical input and comments, E. Loftfield for assistance with the 25-level smoking adjustment variable, and the participants and staff of the UKB, MGBB, and BBJ. The UKB analyses were conducted using applications 7089 and 21552. The authors thank all of the study participants and their families for contributing to the CUB COVID-19 cohort. The genotyping was made possible by the CUB and its COVID-19 Genomics Workgroup members, including A. Califano, W. Chung, C. K. Garcia, D. B. Goldstein, I. Ionita-Laza, K. Kiryluk, R. Mayeux, S. M. O’Byrne, D. Pendrick, M. P. Reilly, S. Sengupta, P. Sims and A.-C. Uhlemann. The authors acknowledge the COVID-19 Host Genetics Initiative consortium for providing infrastructure for collaboration (the members are listed in the Supplementary Information). P.N. is supported by a Hassenfeld Scholar Award from the Massachusetts General Hospital, and grants from the National Heart, Lung, and Blood Institute (R01HL1427, R01HL148565 and R01HL148050). P.N. and B.L.E. are supported by a grant from Fondation Leducq (TNE-18CVD04). S.M.Z. is supported by the NIH National Heart, Lung, and Blood Institute (1F30HL149180-01) and the NIH Medical Scientist Training Program Training Grant (T32GM136651). A.G.B. is supported by a Burroughs Wellcome Fund Career Award for Medical Scientists. G.G. is supported by NIH grant R01 HG006855, NIH grant R01 MH104964, and the Stanley Center for Psychiatric Research. J.P.P. is supported by a John S. LaDue Memorial Fellowship. K.P. is supported by NIH grant 5-T32HL007208-43. P.T.E. is supported by supported grants from the National Institutes of Health (1RO1HL092577, R01HL128914, K24HL105780), the American Heart Association (18SFRN34110082), and by the Foundation Leducq (14CVD01). P.-R.L. is supported by NIH grant DP2 ES030554 and a Burroughs Wellcome Fund Career Award at the Scientific Interfaces. This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, extramural grants from the National Heart, Lung, and Blood Institute, and Fondation Leducq. The opinions expressed by the authors are their own and this material should not be interpreted as representing the official viewpoint of the US Department of Health and Human Services, the National Institutes of Health, or the National Cancer Institute. The CUB is supported by the Vagelos College of Physicians and Surgeons as well as the Precision Medicine Resource and Biomedical Informatics Resource of Irving Institute for Clinical and Translational Research, home of the Columbia University’s Clinical and Translational Science Award (CTSA), funded by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1TR001873.
Author information
Authors and Affiliations
Consortia
Contributions
S.M.Z., S.-H.L., C.W., M.J.M. and P.N. performed statistical modeling of the UKB, FinnGen and MGB data. C.W. collected and analyzed the CUB data. S.M.Z. carried out the analyses of the GWAS and TWAS. P.-R.L. and G.G. carried out the mCA calls. M.J.M. and P.N. supervised the study. S.M.Z. and S.-H.L. drafted the manuscript. All authors critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
P.N. reports grants from Amgen during the conduct of the study and grants from Boston Scientific; grants and personal fees from Apple; personal fees from Novartis and Blackstone Life Sciences; and other support from Vertex outside the submitted work. P.T.E. has received grant support from Bayer AG and has served on advisory boards or consulted for Bayer AG, Quest Diagnostics, MyoKardia and Novartis, outside of the present work. S.M.Z., S.-H.L., M.J.M., G.G., and P.N. have filed a patent application (serial no. 63/079,74) on the prediction of infection from mCAs. G.G. and S.A.M. have filed a patent application (PCT/WO2019/079493) for the MoChA mCA detection method used in the present study. All other authors have no competing interests.
Additional information
Peer review information Nature Medicine thanks Alexander Mentzer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Michael Basson was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 mCA calls by chromosome.
mCA calls by chromosome in the a) MGBB b) FinnGen, and c) CUB. CN-LOH = copy neutral loss of heterozygosity, CUB = Columbia University Biobank, MGBB = Mass-General Brigham Biobank.
Extended Data Fig. 2 Visualization of the diverse range of expanded autosomal mCAs detected across the genome among individuals with a. incident sepsis and b. incident pneumonia in the UKB.
Each point represents one mCA carried by a case, with the x-axis as the chromosome, y-axis as the mCA size in mega-bases of DNA (Mb), color as the copy change, and size of the point as the cell fraction of that mCA. CNN-LOH = copy number neutral loss of heterozygosity, Mb = megabases of DNA, mCA = mosaic chromosomal alterations.
Extended Data Fig. 3 Suggestive associations (P < 0.05) of expanded autosomal mCAs with specific incident infections by Cox proportional-hazards models.
Analyses are adjusted for age, age2, sex, smoking status, and principal components 1-10 of ancestry. Bonferroni correction was used to determine the level of statistical significance (0.05/20 or P < 0.0025). Overall estimates across studies are generated via fixed effect meta-analysis. Error bars show 95% confidence intervals. mCA = mosaic chromosomal alterations.
Extended Data Fig. 4 Associations of a) expanded ChrY and b) expanded ChrX mCAs with incident infections.
Both panels employ Cox proportional-hazards model adjusting for age, age2, sex, smoking status, and principal components 1–10 of ancestry. Error bars show 95% confidence intervals. Bonferroni correction was used to determine the level of statistical significance for each mCA category (P < 0.005). mCA = mosaic chromosomal alterations.
Extended Data Fig. 5
Suggestive associations (P < 0.05) of mCAs with incident infection-related mortality in Biobank Japan Associations of autosomal mCAs with a) organ-system level infections and b) specific infection categories. c) Association of expanded autosomal mCAs with Sepsis. All panels employ Cox proportional-hazards model adjusting for age, age2, sex, smoking status, and principal components 1–10 of ancestry. Error bars show 95% confidence intervals. Bonferroni correction was used to determine the level of statistical significance. Full results are in Supplementary Table 6. Associations are presented among individuals without any cancer history. mCA = mosaic chromosomal alterations.
Extended Data Fig. 6 Incidence rate of at risk population developing each disease (N = 445,101 UKB participants).
95% confidence intervals were calculated based on normal approximation. mCA = mosaic chromosomal alterations.
Extended Data Fig. 7 Associations of expanded mCAs in the UK Biobank with COVID-19 and incident pneumonia.
Associations of expanded mCAs with a. COVID-19 hospitalization across different adjustment models, and b. different COVID-19 phenotypes in fully adjusted logistic regression models. Adjustment models include (1) an unadjusted model, (2) a sparsely adjusted model which adjusts for age, age2, sex, smoking status, and principal components of ancestry, and (3) a fully adjusted model which additionally adjusts for Townsend deprivation index, BMI, and the following comorbidities: Asthma, COPD, CAD, T2D, any cancer, and HTN. Bonferroni correction was used to determine the level of statistical significance. mCA = mosaic chromosomal alterations, COPD = chronic obstructive pulmonary disease, CAD = coronary artery disease, T2D = type 2 diabetes mellitus. c. Association of expanded mCAs with incident pneumonia stratified by sex, adjusted for age, age2, sex (in the All model only), smoking status, and principal components of ancestry. Error bars show 95% confidence intervals. mCA = mosaic chromosomal alterations.
Extended Data Fig. 8 Correlated associations of 63 independent genome-wide significant variants associated with expanded mCAs between different mCA categories in the UKB.
Bonferroni correction was used to determine the level of statistical significance for the correlation analyses (P < 0.05/6 = 0.0083). Across all panels except for panel (a), the labeled genes represent genes attributed to variants that have P < 0.05 across the mCA categories in both axes. mCA = mosaic chromosomal alterations, rp = Pearson correlation.
Extended Data Fig. 9 Association of a mLOY PRS consisting of 156 previously identified20 independent genome-wide significant variants associated with mLOY, with different expanded mCA categories in UKB Females.
Error bands were derived from binomial proportion standard errors. mCA = mosaic chromosomal alterations, mLOY = mosaic Loss-of-chromosome Y, PRS = polygenic risk score.
Extended Data Fig. 10 Pathway enrichment of TWAS results using the Elsevier Pathways.
a. Top results from pathway enrichment analysis of the TWAS results using the Elsevier Pathways. b. Highlighting the GWAS locus-zoom plots for some of the TWAS genes implicated in the top pathways from panel a. Red boxes highlight the gene(s) with strongest association in the TWAS analyses. GWAS = genome-wide association study, TWAS = transcriptome-wide association study.
Supplementary information
Supplementary Information
Supplementary Figs. 1–19 and Supplementary Tables 1–3, 6–12 and 14.
41591_2021_1371_MOESM3_ESM.xlsx
Supplementary Table 4: Infection phenotypic grouping categories across organ systems used in the UK Biobank, MGB Biobank, and Biobank Japan. Supplementary Table 5: Infection phenotypic grouping categories across organ systems used in FinnGen (includes ICD-10, -9, and -8 codes as specified). Supplementary Table 13: 63 independent loci identified in the expanded mCA GWAS. Supplementary Table 15: Transcriptome-wide association using GTExv8-whole blood (n = 670). Supplementary Table 16: Gene set pathway enrichment analysis of the transcriptome-wide analyses (from Supplementary Data 4) using the Elsevier Pathway Collection through EnrichR. Supplementary Table 17: Other phenotype definitions used in the UK Biobank COVID-19 sensitivity analyses. Supplementary Table 18: Other phenotype definitions used in MGB Biobank. Supplementary Table 19: Other phenotype definitions used in FinnGen.
Rights and permissions
About this article

Cite this article
Zekavat, S.M., Lin, SH., Bick, A.G. et al. Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection. Nat Med 27, 1012–1024 (2021). https://doi.org/10.1038/s41591-021-01371-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01371-0
This article is cited by
-
Clonal haematopoiesis, ageing and kidney disease
Nature Reviews Nephrology (2024)
-
Tissue mosaicism following stem cell aging: blood as an exemplar
Nature Aging (2024)
-
Genetic variation across and within individuals
Nature Reviews Genetics (2024)
-
Clonal hematopoiesis and its impact on the aging osteo-hematopoietic niche
Leukemia (2024)
-
Clonal chromosomal mosaicism and loss of chromosome Y in elderly men increase vulnerability for SARS-CoV-2
Communications Biology (2024)
