
Abstract
The Zero Childhood Cancer Program is a precision medicine program to benefit children with poor-outcome, rare, relapsed or refractory cancer. Using tumor and germline whole genome sequencing (WGS) and RNA sequencing (RNAseq) across 252 tumors from high-risk pediatric patients with cancer, we identified 968 reportable molecular aberrations (39.9% in WGS and RNAseq, 35.1% in WGS only and 25.0% in RNAseq only). Of these patients, 93.7% had at least one germline or somatic aberration, 71.4% had therapeutic targets and 5.2% had a change in diagnosis. WGS identified pathogenic cancer-predisposing variants in 16.2% of patients. In 76 central nervous system tumors, methylome analysis confirmed diagnosis in 71.1% of patients and contributed to a change of diagnosis in two patients (2.6%). To date, 43 patients have received a recommended therapy, 38 of whom could be evaluated, with 31% showing objective evidence of clinical benefit. Comprehensive molecular profiling resolved the molecular basis of virtually all high-risk cancers, leading to clinical benefit in some patients.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
WGS, RNAseq and methylation data generated by this study are available from the European Genome-phenome Archive under accession number EGAS00001004572. Databases used to help filter, prioritize and interpret variants are available online, including COSMIC (https://cancer.sanger.ac.uk/cosmic), Cancer Gene Census (https://cancer.sanger.ac.uk/census), Pecan (https://pecan.stjude.cloud/), dbscSNV (http://www.liulab.science/dbscsnv.html), dbNSFP (https://sites.google.com/site/jpopgen/dbNSFP), ExAC (http://exac.broadinstitute.org/), gnomAD (https://gnomad.broadinstitute.org/), MGRB (https://sgc.garvan.org.au/), GIAB (https://jimb.stanford.edu/giab-resources), Platinum Genomes (https://github.com/Illumina/PlatinumGenomes), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), ESP (https://evs.gs.washington.edu/EVS/) and 1000 Genomes (https://www.internationalgenome.org/data).
Code availability
Software and scripts related to this publication are available at https://github.com/CCICB/2020-hrPC-landscape.
References
Steliarova-Foucher, E. et al. Changing geographical patterns and trends in cancer incidence in children and adolescents in Europe, 1991–2010 (Automated Childhood Cancer Information System): a population-based study. Lancet Oncol. 19, 1159–1169 (2018).
Gu, Z. et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 51, 296–307 (2019).
Stewart, E. et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell 34, e419 (2018).
Northcott, P. A. et al. The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317 (2017).
Pugh, T. J. et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284 (2013).
Berger, M. F. & Mardis, E. R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 15, 353–365 (2018).
Jones, D. T. W. et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat. Rev. Cancer 19, 420–438 (2019).
Kurzrock, R. & Giles, F. J. Precision oncology for patients with advanced cancer: the challenges of malignant snowflakes. Cell Cycle 14, 2219–2221 (2015).
Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376 (2018).
Grobner, S. N. et al. The landscape of genomic alterations across childhood cancers. Nature 555, 321–327 (2018).
Parsons, D. W. et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2, 616–624 (2016).
Chang, W. et al. Multidimensional clinomics for precision therapy of children and adolescent young adults with relapsed and refractory cancer: a report from the center for cancer research. Clin. Cancer Res. 22, 3810–3820 (2016).
Harttrampf, A. C. et al. Molecular screening for cancer treatment optimization (MOSCATO-01) in pediatric patients: a single-institutional prospective molecular stratification trial. Clin. Cancer Res. 23, 6101–6112 (2017).
Khater, F. et al. Molecular profiling of hard-to-treat childhood and adolescent cancers. JAMA Netw. Open 2, e192906 (2019).
Mody, R. J. et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA 314, 913–925 (2015).
Worst, B. C. et al. Next-generation personalised medicine for high-risk paediatric cancer patients - the INFORM pilot study. Eur. J. Cancer 65, 91–101 (2016).
Allen, C. E. et al. Target and agent prioritization for the children’s oncology group-National Cancer Institute Pediatric MATCH trial. J. Natl Cancer Inst. 109, djw274 (2017).
Parsons, D. W. et al. Identification of targetable molecular alterations in the NCI-COG Pediatric MATCH trial. J. Clin. Oncol. 37, 10011–10011 (2019).
Priestley, P. et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019).
Campbell, P. J., Getz, G., Stuart, J. M., Korbel, J. O. & Stein, L. D. -cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Campbell, B. B. et al. Comprehensive analysis of hypermutation in human cancer. Cell 171, e1010 (2017).
Davies, H. et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 23, 517–525 (2017).
Vaske, O. M. et al. Comparative tumor RNA sequencing analysis for difficult-to-treat pediatric and young adult patients with cancer. JAMA Netw. Open 2, e1913968 (2019).
McFarland, J. M. et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat. Commun. 9, 4610 (2018).
Rodon, J. et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat. Med. 25, 751–758 (2019).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Van Allen, E. M., Wagle, N. & Levy, M. A. Clinical analysis and interpretation of cancer genome data. J. Clin. Oncol. 31, 1825–1833 (2013).
Hoskinson, D. C., Dubuc, A. M. & Mason-Suares, H. The current state of clinical interpretation of sequence variants. Curr. Opin. Genet. Dev. 42, 33–39 (2017).
Griffith, M. et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 49, 170–174 (2017).
Drilon, A. et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 378, 731–739 (2018).
Ziegler, D. S. et al. Brief report: potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br. J. Cancer 119, 693–696 (2018).
Bell, R. J. et al. Understanding TERT promoter mutations: a common path to immortality. Mol. Cancer Res. 14, 315–323 (2016).
Rheinbay, E. et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 (2020).
Jouhilahti, E. M. et al. The human PRD-like homeobox gene LEUTX has a central role in embryo genome activation. Development 143, 3459–3469 (2016).
Gough, S. M., Slape, C. I. & Aplan, P. D. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 118, 6247–6257 (2011).
Stathis, A. & Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24 (2018).
Zimmerman, M. W. et al. MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 8, 320–335 (2018).
Cameron, D. L., et al. GRIDSS, PURPLE, LINX: unscrambling the tumor genome via integrated analysis of structural variation and copy number. Preprint at bioRxiv https://www.biorxiv.org/content/10.1101/781013v1 (2019).
Bailey, M. L. et al. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol. Cancer Ther. 13, 724–732 (2014).
Leung, J. W. et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 31, 260–274 (2017).
Jones, D. T. W. et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 68, 8673–8677 (2008).
Kohashi, K. et al. Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression: three subtypes of rhabdoid tumor according to their histological features. Mod. Pathol. 29, 1232–1242 (2016).
Donson, A. M. et al. Unique molecular characteristics of radiation-induced glioblastoma. J. Neuropathol. Exp. Neurol. 66, 740–749 (2007).
Zhang, J., Nichols, K. E. & Downing, J. R. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med. 374, 1391 (2016).
Nielsen, M. et al. Survival of MUTYH-associated polyposis patients with colorectal cancer and matched control colorectal cancer patients. J. Natl Cancer Inst. 102, 1724–1730 (2010).
Shlien, A. et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat. Genet. 47, 257–262 (2015).
Wimmer, K. & Etzler, J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum. Genet. 124, 105–122 (2008).
Haradhvala, N. J. et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 9, 1746 (2018).
Harris, M. H. et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the individualized cancer therapy (iCat) study. JAMA Oncol. 2, 608–615 (2016).
Marks, L. J. et al. Precision medicine in children and young adults with hematologic malignancies and blood disorders: the Columbia University experience. Front. Pediatr. 5, 265 (2017).
Pincez, T., et al. Feasibility and clinical integration of molecular profiling for target identification in pediatric solid tumors. Pediatr. Blood Cancer 64 https://doi.org/10.1002/pbc.26365 (2017).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Fabregat, A. et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 44, D481–D487 (2016).
Jassal, B. et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 48, D498–D503 (2020).
Khaw, S. L. et al. Venetoclax responses of pediatric ALL xenografts reveal sensitivity of MLL-rearranged leukemia. Blood 128, 1382–1395 (2016).
Villalobos-Ortiz, M., Ryan, J., Mashaka, T. N., Opferman, J. T. & Letai, A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 27, 999–1007 (2019).
Blombery, P. et al. Characterization of a novel venetoclax resistance mutation (BCL2 Phe104Ile) observed in follicular lymphoma. Br. J. Haematol. 186, e188–e191 (2019).
Guieze, R. et al. Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid malignancies. Cancer Cell 36, e313 (2019).
Lee, D. P., Skolnik, J. M. & Adamson, P. C. Pediatric phase I trials in oncology: an analysis of study conduct efficiency. J. Clin. Oncol. 23, 8431–8441 (2005).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv https://arxiv.org/abs/1303.3997 (2013).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.1–11.10.33 (2013).
Gayevskiy, V., Roscioli, T., Dinger, M. E. & Cowley, M. J. Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics 35, 122–125 (2019).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Paila, U., Chapman, B. A., Kirchner, R. & Quinlan, A. R. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol. 9, e1003153 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Pinese, M. et al. The medical genome reference bank contains whole genome and phenotype data of 2570 healthy elderly. Nat. Commun. 11, 435 (2020).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31–31 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Cameron, D. L. et al. GRIDSS: sensitive and specific genomic rearrangement detection using positional de Bruijn graph assembly. Genome Res. 27, 2050–2060 (2017).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag, 2016).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Craig, D. W. et al. A somatic reference standard for cancer genome sequencing. Sci. Rep. 6, 24607 (2016).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, e524 (2019).
Cheng, J. et al. MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol. 20, 48 (2019).
Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
Signal, B., Gloss, B. S., Dinger, M. E. & Mercer, T. R. Machine learning annotation of human branchpoints. Bioinformatics 34, 920–927 (2018).
Xiong, H. Y. et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806 (2015).
Garrido-Martin, D., Palumbo, E., Guigo, R. & Breschi, A. ggsashimi: Sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput. Biol. 14, e1006360 (2018).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Fu, W. et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220 (2013).
Fairley, S., Lowy-Gallego, E., Perry, E. & Flicek, P. The international genome sample resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Res. 48, D941–D947 (2019).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Vaser, R., Adusumalli, S., Leng, S. N., Sikic, M. & Ng, P. C. SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 (2016).
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65 (2013).
Kim, S., Jhong, J. H., Lee, J. & Koo, J. Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 10, 2 (2017).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 24, 2125–2137 (2015).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Sondka, Z. et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Haas, B. J. et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 20, 213 (2019).
Davidson, N. M., Majewski, I. J. & Oshlack, A. JAFFA: high sensitivity transcriptome-focused fusion gene detection. Genome Med. 7, 43 (2015).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
O, J. H., Lodge, M. A. & Wahl, R. L. Practical PERCIST: a simplified guide to PET response criteria in solid tumors 1.0. Radiology 280, 576–584 (2016).
Wen, P. Y. et al. Response assessment in neuro-oncology clinical trials. J. Clin. Oncol. 35, 2439–2449 (2017).
Acknowledgements
We sincerely thank patients and parents for participating in this study. We thank the many clinicians and health professionals for their time acquiring consenting patients and for collection and coordination of samples and associated clinical data at Sydney Children’s Hospital, Randwick; The Children’s Hospital at Westmead; the John Hunter Hospital; the Queensland Children’s Hospital; the Royal Children’s Hospital Melbourne; the Monash Children’s Hospital; the Adelaide Women’s & Children’s Hospital; and the Perth Children’s Hospital. We thank M. Weber (Prince of Wales Hospital) for assistance with anatomical pathology expert input. The authors thank the Sydney Children’s Tumour Bank Network for providing samples and related clinical information for this study. We thank the staff of the Personalised Medicine Theme of the Children’s Cancer Institute for their dedicated work on Zero Childhood Cancer. We thank M. Dinger, B. Lundie, J. Stockmeyer, A. Statham, J. Copty and the staff at the Kinghorn Centre for Clinical Genomics for helpful discussions related to, and rapid processing of, WGS; J. Mattick and M. Dziadek from the Garvan Institute for helpful discussions related to WGS and for supporting fundraising initiatives; S. Eggers and the staff at the Murdoch Children’s Research Institute/Victorian Comprehensive Genetics Service for rapid processing of RNAseq; and the staff at the Australian Genome Research Facility for rapid processing of methylation data. We thank the team from the National Computational Infrastructure, which is supported by the Australian Government. We thank G. Asimenos, D. Pollard and the team from DNAnexus for support with genomic data processing and helpful discussions regarding pipeline development. We thank the Australian Federal Government Department of Health, the New South Wales State Government and the Australian Cancer Research Foundation for funding to establish infrastructure to support the Zero Childhood Cancer personalized medicine program. We thank the Kids Cancer Alliance, Cancer Therapeutics Cooperative Research Centre, for supporting the development of a personalized medicine program; Tour de Cure for supporting tumor biobank personnel; The Steven Walter Children’s Cancer Foundation and The Hyundai Help 4 Kids Foundation for supporting G.M.M. and P.G.E.; and the Lions Kids Cancer Genome Project, a joint initiative of Lions International Foundation, the Australian Lions Children’s Cancer Research Foundation (ALCCRF), the Garvan Institute of Medical Research, the Children’s Cancer Institute and the Kids Cancer Centre, Sydney Children’s Hospital. Lions International and ALCCRF provided funding to perform WGS and for key personnel, with thanks to J. Collins for project governance and advocacy. We thank the Cure Brain Cancer Foundation for supporting RNA sequencing of patients with brain tumors; the Kids Cancer Project for supporting molecular profiling and molecular and clinical trial personnel; and the University of New South Wales, W. Peters and the Australian Genomics Health Alliance for providing personnel funding support. The New South Wales Ministry of Health-funded Luminesce Alliance provided funding support for computational personnel and infrastructure. The Medical Research Future Fund, the Australian Brain Cancer Mission, the Minderoo Foundation’s Collaborate Against Cancer Initiative and funds raised through the Zero Childhood Cancer Capacity Campaign, a joint initiative of the Children’s Cancer Institute and the Sydney Children’s Hospital Foundation, supported the national clinical trial and associated clinical and research personnel. We thank the Kinghorn Foundation for personnel support. We thank the National Health and Medical Research Council of Australia (fellowships APP1059804 and APP1157871 to R.B.L), the Cancer Institute of New South Wales and New South Wales Health (fellowship funding for M.J.C.). We thank the 2018 Priority-Driven Collaborative Cancer Research Scheme, co-funded by Cancer Australia and My Room, for personnel and computational support (grant no. 1165556 awarded to M.J.C.). Zero Childhood Cancer is a joint initiative led by the Children’s Cancer Institute and Sydney Children’s Hospital, Randwick.
Author information
Authors and Affiliations
Contributions
M.H., G.M.M. and M.D.N. conceived of and designed the Zero Childhood Cancer project. V.T. and E.V.A.M. led and managed the Zero Childhood Cancer Program. D.S.Z., T.O., F. Alvaro, G.B.M., L.D.P., N.G.G., H.T., P.W., S.-L.K., J.R.H. and A.S.M. were clinical leads at recruiting centers and coordinated patient selection. D.G.-W., P.A.S. and E.M. checked sample quality and identity and prepared samples for sequencing. A.J.G. provided pathology analysis. M.W., C.M., M.P., A.K., E.E.W., P.S., R.B.-J., M.S., I.M., F. Abascal, J.B., P.P., M.E.G., V.G. and M.J.C. developed bioinformatic methods and performed the computational genomics analyses. J.K., K.T., M.W., N.A.B. and M.P. interpreted and reported germline findings. L.M.S.L., D.-A.K.-Q., P.B., A.S., M.E.M.D., E.D.G.F., E.V.A.M., P.G.E. and M.J.C. analyzed and curated molecular findings. L.M.S.L., D.-A.K.-Q., T.N.T., G.M.M. and D.S.Z. led the Multidisciplinary Tumor Board discussions and recommendations. L.M.S.L., D.-A.K.-Q. and D.Z. evaluated patient response data. M.D.N., T.N.T. and R.B.L. provided childhood cancer expertise and advice. D.M.T. contributed precision medicine expertise and advice. P.G.E. and M.J.C. conceived of, designed and wrote the manuscript, with comments and contributions from all authors, particularly M.P. who wrote the germline findings section and L.L. and D.S.Z. who wrote the clinical impact sections.
Corresponding authors
Ethics declarations
Competing interests
All authors, except P.G.E., S.-L.K. and D.S.Z., declare that they have no competing interests as defined by Nature Research or other interests that might be perceived to influence the interpretations of the article. D.S.Z. has received funding for travel and consulting from Bayer. P.G.E. and S.-L.K. are recipients of a share in milestone and royalty payments paid to the Walter and Eliza Hall Institute of Medical Research for the development of venetoclax.
Additional information
Peer review information Javier Carmona was the primary editor on this article, and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 ZERO Workflow.
a, The workflow of samples through the ZERO program is shown. Patients were enrolled at one of eight paediatric hospitals, samples processed centrally then profiled at various national centres of excellence for WGS, RNAseq and methylome analysis. The data from each molecular profiling platform are analysed centrally and integrated where possible via dedicated bioinformatics pipelines. The resulting molecular aberrations or classifications from all analysis pipelines are collated and stored in a relational database and integrated by an in-house integration system, Glooee. Here, Glooee applies algorithms that score and rank each variant for prioritisation and generates visualisations to support data quality control assessment and downstream curation. The curation process is critical for classifying molecular aberrations prior to reporting changes to the Molecular Tumour Board (MTB). The multidisciplinary curation team determines the pathogenicity of each variant (C1–C5) consistent with published guidelines29. Ultimately the molecular aberrations are assessed for their reportability (that is of potential interest to clinicians), potential targetability with anticancer drugs, potential to support, refine, or propose a change of diagnosis, prognosis, or indicate the presence of a germline cancer predisposition syndrome. The MTB determines the strengths of molecular, preclinical or clinical data supporting potential therapeutic findings. Ultimately, the MTB seeks consensus on what is reported back to the patient. b, the variant types for which each sequencing platform was used. The red colouring in Tumour WGS and RNAseq indicates that both platforms were used to corroborate these variant types where possible.
Extended Data Fig. 2 Mutational Landscape of High-Risk Paediatric Cancers.
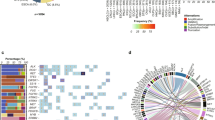
An oncoprint of the reportable germline and somatic events, of any type, observed in more than 2 patients. Genes are classified into broad pathway categories and ordered by the percentage of patients harbouring an alteration in the specified gene. The barplot above shows the number of events in each patient highlighted by aberration type. The barplot on the right shows the total number of events in each indicated gene, highlighted by aberration type and represents genes where more than one aberration, for example CNV gain and over-expression, occurred in the same sample. Where individual genes are shown for each patient, the colour represents the variant type (legend shown on right). Where these bars have multiple colours, it is because the same variants were detected by different techniques, or there are multiple variant types affecting each allele. The heatmaps on the bottom show the distribution of cancer type, stage of disease and sex of patient (legend shown on the right).
Extended Data Fig. 3 Pathogenic Aberrations in High-Risk Paediatric Cancers.
a, Genes recurrently altered by any of the different variant types are shown, grouped by broad pathway categories and ordered by the frequency of the mutation. The plot includes all genes affected in >2 patients. The colouring of the bars indicates the proportion of each variant type. b–f, Circos plots of the genome-wide copy number profiles for each major category of cancers. The amplifications and deletions or LOH relative to the tumour ploidy are shown according to the inset legend. The scale on both rings is 0–70% and inverted for the inner ring. The genes most frequently observed with high-level amplifications, homozygous deletions and immunoglobulin or T-cell receptor gene rearrangements (*) are shown. The number of patients in each subtype are CNS (n=92), HM (n=43), NBL (n=20; n=8 males), Sarcoma (n=61), other solid tumours (n=35).
Extended Data Fig. 4 Complex structural rearrangements driving novel fusions.
a, BRD4-LEUTX fusion identified from RNAseq and WGS from a CNS embryonal tumour at diagnosis in an infant. This figure reveals that at least one SV joined exons 1–11 of BRD4 to exons 2–3 of LEUTX. These exons were highly expressed (coverage histogram above each exon) and the resulting fusion was in-frame and retained the bromodomains in BRD4 and homeobox domain in LEUTX. The expression of LEUTX across cohort is depicted (right), with patient highlighted in red, other CNS embryonal tumours in green and cohort in black. b, LINX visualisation guide, highlighting how multiple SV can be chained together into a derivative chromosome, where SV breakpoints flank defined CNV segments. c, Complex genome topology underlies an out-of-frame TP53-SUZ12 fusion identified from an MPNST tumour at initial diagnosis in an adolescent. The plot shows a predicted derivative chromosome, characterised by many SVs (purple lines) and CNV segments (predominantly green segments with ploidy of 2–4) of 10 chromosomes as indicated. d, A dot plot representation of expression TPM values of TP53 in the cohort, patient with TP53-SUZ12 highlighted in red, other MPNST in green and cohort in black, black horizontal line signifies cohort mean TPM. e, A pathogenic 607 bp deletion affecting intron 1, a 96 Kb deletion and a 66 Kb inversion, both affecting exons 2–3 of IKZF1 were identified in three patients with pre-B ALL. The inversion was likely in trans with a germline pathogenic splicing variant (IKZF1:c.40+1G>A). In two other pre-B ALL cases a tumour with a haploid genome had an IKZF1 deletion and another had a deletion and a likely pathogenic somatic variant (IKZF1:c.544T>C, p.(Cys182Arg)); in both cases the CNV was not deemed to be a driver mutation and was not reported. The plot was made using GenomePaint (X Zhou, in prep).
Extended Data Fig. 5 Integration of WGS and RNAseq reveals the impact of mutations on splicing.
a–l, The impact of unique somatic mutations on gene splicing, where details about each mutation are in Supplementary Table 3. In each case, a patient’s mutation is shown in red, relative to the exon-intron boundary. The patient’s RNAseq read coverage is shown above (red) as a histogram, and the number of reads supporting each splice junction, compared to four cancer-type matched controls below (purple).
Extended Data Fig. 6 Methylation classification results from CNS tumours.
CNS tumour methylation array classification results from 76 patients were binned into those where a strong, weak or no match was made to a known tumour subtype and then by whether that classification matched, differed, or resulted in a change of the initial diagnosis determined from standard of care diagnostic testing.
Extended Data Fig. 7 Tumour Mutation Burden across the cohort.
Somatic single nucleotide tumour mutation burden (TMB) for each patient and the mean per cancer type (red line), expressed as genome-wide mutations per MB. Ninety-four percent of tumours had low TMB (<10 mutations per MB), consistent with previous reports10.
Extended Data Fig. 8 Germline and somatic features of ultramutated CNS tumours.
a, Circos plot depicting the genome-wide mutation profile of an HGG, from a child, with 165 mutations/MB. The inner area shows SVs, followed by the minor allele ploidy (here highlighting the expected loss of heterozygosity on chrX), then CNV (deletions in red, amplifications in green), followed by somatic SNVs coloured according to the figure legend. b, Mutation signature analysis69 (Methods) showed that most mutations matched previously known mutation signatures. c, The burden of each mutation signature is shown, where signature 14 has been associated with CMMRD and somatic POLE/POLD1 mutations in the exonuclease domain49. A somatic POLE hotspot mutation (p.Leu424Val) was identified, which was classified as a VUS in ClinVar (RCV000033144) at the time of sequencing. d, MLPA analysis revealed a heterozygous exon 12 duplication but could not resolve the breakpoints. We re-inspected the WGS data which revealed the heterozygous exon 12 duplication of PMS2 from 7:6,021,129–6,023,032 (POLE:p.(Lys670Ala725dup). None of our SV detection tools from WGS or RNAseq identified this variant. Exons 12–15 of PMS2 are difficult to sequence due to 99% sequence homology with a pseudogene. WGS easily identified the other pathogenic allele (c.949C>T, p.(Gln317Ter)) and loss of PMS2 expression confirmed by IHC, resulting in a diagnosis of CMMRD. Panels E-H similarly represent the molecular findings from a hemispheric HGG tumour with 353 mutations/MB. In this case, signature 14 again prompted us to look for POLE/POLD1 variants, revealing a clear somatic deletion of one POLE allele and a subclonal hotspot mutation (p.S459F) with just 4 supporting reads and several VUS on the remaining allele. RNAseq did not support the expression of this hotspot mutation, so it was not reported. Both examples highlight how somatic features of the tumour, in this case TMB and mutation signatures prompted a deeper investigation into the molecular drivers of these tumours.
Extended Data Fig. 9 Germline HR mutation driving a tumour with signature 3.
An example of concordant germline and somatic findings supporting a treatment recommendation, showing the tumour’s observed mutation spectrum (top panel), the HR-associated signature 3 mutation spectrum (middle panel), and the inferred signature exposure in the tumour (bottom panel), with signature 3 the dominant signal supporting the recommendation of a PARP inhibitor.
Extended Data Fig. 10 Multi-platform approach in ZERO increases reportable events.
a, The number of reportable events identified in each individual patient, coloured by the source of the data, whether from WGS only, RNAseq only, both WGS and RNA, germline WGS or by methylation array. The x-axis is each patient in the cohort. The y-axis is the number of reportable findings in each patient. b, The total number of reportable variants separated by type of event and coloured by the platform identifying the event. SNV: Single Nucleotide Variant, CNV: Copy Number Variant, SV: Structural Variant, EXP: Gene Expression, GL: Germline, METH: Methylation, TMB: Tumour Mutation Burden.
Supplementary information
Supplementary Information
Supplementary Table legends 1–5 and Supplementary Tables 2 and 4.
Supplementary Table
Supplementary Tables 1, 3 and 5.
Rights and permissions
About this article

Cite this article
Wong, M., Mayoh, C., Lau, L.M.S. et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 26, 1742–1753 (2020). https://doi.org/10.1038/s41591-020-1072-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-020-1072-4
This article is cited by
-
Molecular and clinicopathologic characteristics of CNS embryonal tumors with BRD4::LEUTX fusion
Acta Neuropathologica Communications (2024)
-
CNS embryonal tumour with concomitant novel BRD4::CTRC1 fusion and BCOR internal tandem duplication – evidence for synergism and non-mutually exclusive alterations in CNS embryonal tumours
Acta Neuropathologica Communications (2024)
-
The evolutionary impact of childhood cancer on the human gene pool
Nature Communications (2024)
-
Prognostic DNA mutation and mRNA expression analysis of perineural invasion in oral squamous cell carcinoma
Scientific Reports (2024)
-
Functional precision medicine for pediatric cancers
Nature Medicine (2024)
