Article Text

Abstract
Objective To investigate whether the Ala143Thr variant of the α-galactosidase A gene (A143T/GLA), with conflicting interpretations of pathogenicity, is associated with Fabry cardiomyopathy.
Methods The index patient, a woman in her 60s with cardiomyopathy, was screened for variants in 59 cardiomyopathy-related genes. A143T/GLA, the only rare variant found, was screened in 10 relatives. GLA activity and lyso-Gb3 levels were measured and echocardiography was performed in 8 of 9 subjects carrying A143T/GLA. Cardiac magnetic resonance (CMR) imaging and 18F-fluorodeoxyglucose (FDG) positron emission tomography/CT (PET/CT) were performed in four adult A143T/GLA carriers. Endomyocardial biopsy was obtained from two adult A143T/GLA carrying sons of the index patient.
Results The index patient and her elder son had a pacemaker implantation because of sick sinus syndrome and atrioventricular block. GLA activities were decreased to 25%–40% of normal in both sons and one granddaughter. Lyso-Gb3 levels were elevated in both sons. In CMR, the index patient and her two sons had left ventricular (LV) hypertrophy and/or dilatation. The elder son had late gadolinium enhancement, high CMR-derived T1 time and positive FDG signal in PET/CT in the basal inferolateral LV wall. The younger son had low T1 time and the mother had positive FDG signal in PET/CT in the basal inferolateral LV wall. Endomyocardial biopsy of both sons showed myocardial accumulation compatible with glycolipids in light and electron microscopy, staining with anti-Gb3 antibody available for the younger son. Five female relatives with A143T/GLA had no cardiomyopathy in cardiac imaging.
Conclusions A143T/GLA is likely a late-onset Fabry cardiomyopathy causing variant with incomplete penetrance.
- familial cardiomyopathies
- metabolic heart disease
- clinical genetics
- cardiac magnetic resonance (CMR) imaging
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- familial cardiomyopathies
- metabolic heart disease
- clinical genetics
- cardiac magnetic resonance (CMR) imaging
Introduction
Fabry disease (FD) is a rare X-chromosome linked lysosomal storage disorder caused by mutations in the α-galactosidase A gene (GLA). According to Human Gene Mutation Database, over 900 mutations GLA have been reported worldwide. GLA mutations result in functionally deficient GLA enzyme, which leads to progressive accumulation of glycosphingolipid substrates, particularly globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) in different organs.1 In the late-onset form of FD, which is more common than the classic type, patients have residual enzyme activity. Cardiomyopathy is often the predominant or the only manifestation of the late-onset disease, and typically develops in the middle age in hemizygous and heterozygous subjects.1 Cardiovascular complications are the major cause of death in FD.2 Enzyme replacement therapy (ERT) is an effective treatment for FD when started before permanent organ damage develops.3
Genetic analyses are currently widely used in the diagnosis of cardiomyopathies.4 In a large cohort of European patients with hypertrophic cardiomyopathy, GLA mutations accounted for 0.5% of cases.5 In other studies, 3%–6.3% of males with hypertrophic cardiomyopathy were diagnosed with FD.6 The pathogenicity and clinical importance of all GLA variants, however, is not clear. Particularly, the missense variant Ala143Thr of GLA (c.427G>A; A143T/GLA), common in the newborn screening in some areas of the USA and patient populations with FD, has been considered pathogenic in several studies but benign in others.7–18
In the present study, we describe a Finnish family with A143T/GLA and cardiomyopathy. Our aim was to investigate the association of A143T/GLA with cardiomyopathy in the family, and to examine by clinical, biomarker, cardiac imaging and histological methods if cardiomyopathy in family members is compatible with Fabry cardiomyopathy.
Methods
Study design
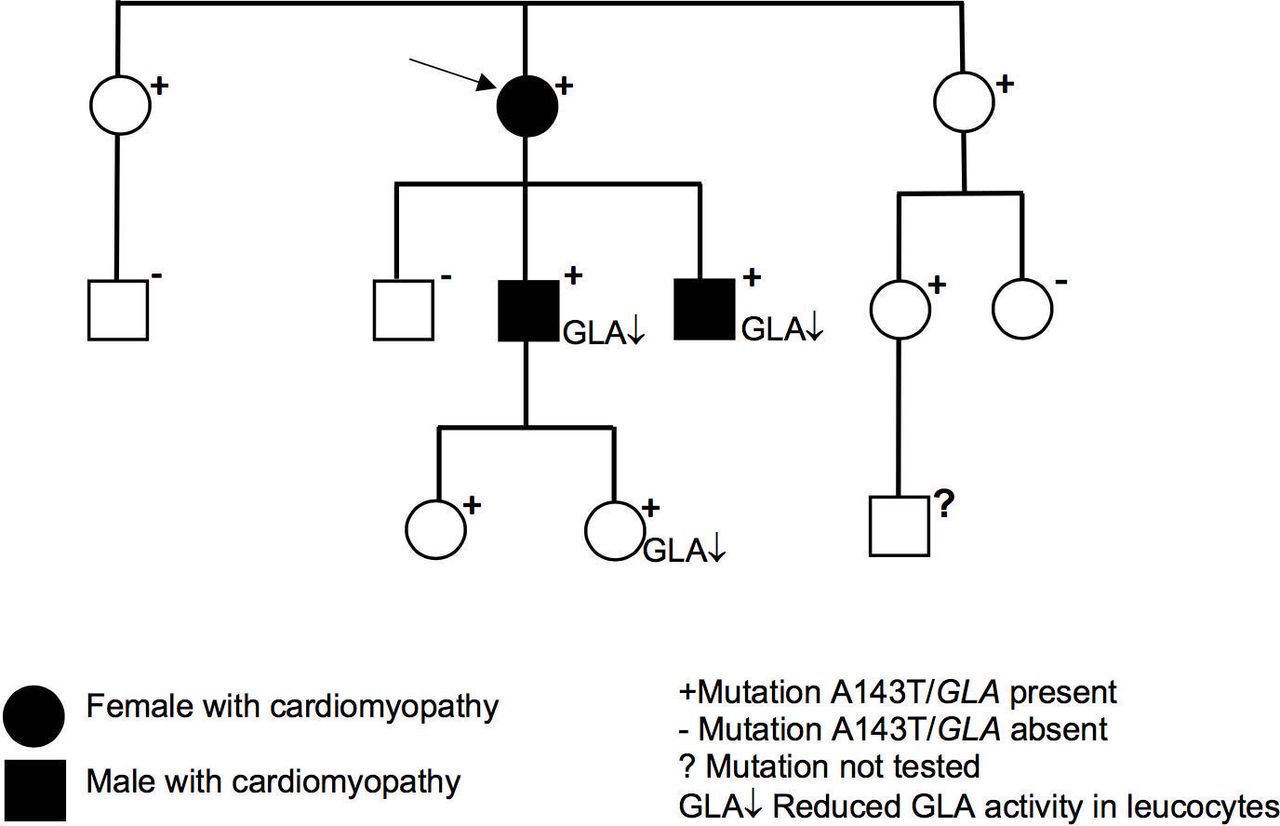
The present family study started with two members of a Finnish family from the Kuopio University Hospital area, who had cardiomyopathy, and included investigation of altogether 11 family members (figure 1).

The family tree of the index patient (arrow) carrying Ala143Thr variant of the α-galactosidase A gene (A143T/GLA). Of 11 family members tested, 8 were carriers of A143T/G LA. Both A143T/G LA-positive males and a young female had decreased levels of G LA activity in leucocytes. Cardiomyopathy was diagnosed in the female index patient in her 60s, her son in his 30s and her son in his 20s.
Genetic analysis of 59 cardiomyopathy-related genes
Genetic analysis was performed in the Genome Center of the University of Eastern Finland. The genetic screening from the DNA of the index patient and his two sons with cardiomyopathy covered coding regions of the 59 genes related to cardiomyopathy, including GLA. Cascade screening of A143T/GLA was performed with Sanger sequencing in all available relatives (n=10). For details see online supplementary information.
Supplemental material
In silico structural analysis of mutated GLA protein
Molecular structure of A143T/GLA mutated protein was obtained using in silico structural analysis. For details see online supplementary information.
GLA enzyme activity and lyso-Gb3 levels
GLA enzyme activity and lyso-Gb3 levels were measured in available A143T/GLA-positive subjects (n=7). For details see online supplementary information.
Clinical examination
Family members with A143T/GLA living in Finland (n=7) were examined at the Heart Center of the Kuopio University Hospital (n=6) or at the Heart Hospital of the Tampere University Hospital (n=1) according to Finnish FD protocol.19 A paediatrician examined the children (n=2), and a cardiologist (KV) and an internist examined the adults (n=5). The standard 12-lead ECG was recorded in all adult subjects. Examinations by an ophthalmologist and dermatologist were performed, when clinically relevant. One elderly female A143T/GLA carrier living in Sweden was diagnosed not to have FD, but no detailed information on her clinical, biomarker and imaging findings is available.
Echocardiography
Cardiac ultrasound examinations were recorded by a cardiologist in adult carriers of A143T/GLA (n=5) and by a paediatric cardiologist in children (n=2). Echocardiography included two-dimensional echocardiography using a GE Vivid Q Ultrasound equipment. Conventional echocardiographic parameters were measured according to current guidelines. Left ventricular hypertrophy (LVH) was defined as maximal left ventricular (LV) wall thickness ≥13 mm in diastole.
Cardiac MRI
Cardiac magnetic resonance (CMR) imaging was performed in all available adult A143T/GLA carriers (n=4) by imaging cardiologist (MH) and radiologist (LL-R) by using 1.5 T full-body scanner (Magnetom AERA, Siemens Healthcare, Erlangen, Germany). One adult with normal ultrasound refused CMR. In children (n=2) only cardiac ultrasound was recorded. CMR included cine imaging, late gadolinium enhancement (LGE) images and image analysis. Non-contrast myocardial T1 mapping was available in both sons of the index patient. For details see online supplementary information.
18F-fluorodeoxyglucose positron emission tomography/CT
All available adult A143T/GLA carriers (n=4) underwent 18F-fluorodeoxyglucose (FDG) positron emission tomography/CT (PET/CT). PET/CT was used to characterise cardiac metabolic activity and to monitor treatment response in cardiac glucose uptake. Maximum standardised uptake value (SUV) was determined. To calculate the metabolic volumes of abnormal FDG uptake, a threshold of SUV 2.7 was used. PET scanning results were analysed by clinical physiologists (JN-Q, TL). For details see online supplementary information.
Endomyocardial biopsy
Endomyocardial biopsy was performed in two A143T/GLA-positive sons of the index patient, who had signs of cardiomyopathy. Myocardial specimens for histological analysis, immunohistochemistry and electron microscopy were obtained. Several representative biopsies were taken. Myocardial specimens were stained for light and transmission electron microscopy in both sons. Immunohistochemical staining with an anti-Gb3 antibody was available in the younger son. Specimen were analysed in the Diagnostic Imaging Center of Kuopio University Hospital by a senior cell biologist (AN). For details see online supplementary information.
Results
Genetic findings
In the genetic screening of the index patient in her 60s, her son in his 30s and the son in his 20s with cardiomyopathy, A143T/GLA was found. No other pathogenic or likely pathogenic variants were identified in 59 cardiomyopathy-related genes. In the cascade genetic screening, five additional relatives, all females aged from 7 to 69 years, carried A143T/GLA ( figure 1).
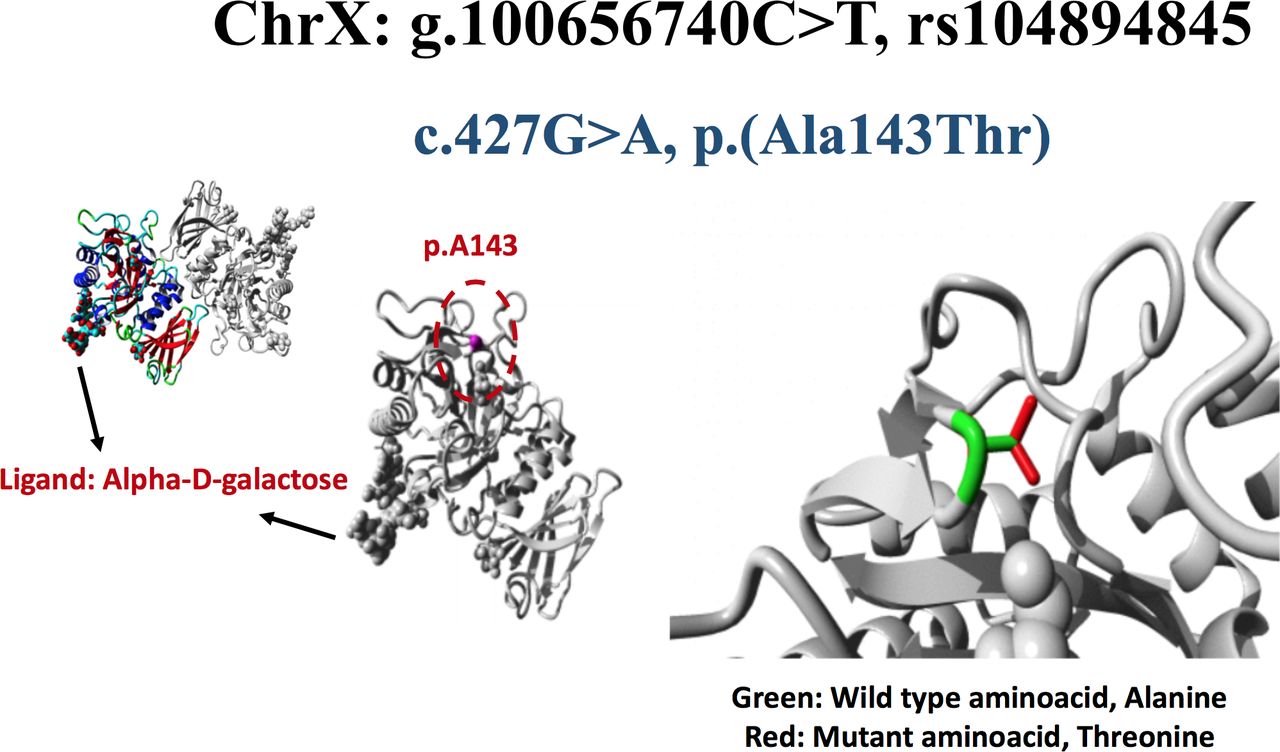
Molecular structure of A143T mutated GLA protein
The residue is located on the surface of the GLA protein. The mutant residue (threonine) is larger than the residue of the wild-type. Mutation of this residue can disturb interactions with other molecules (figure 2).

Structural analysis of GLA missense variant, c.427G>A, p.Ala143Thr. The wild-type residue is Ala143, shown as magenta coloured ball on the surface of GLA protein. Ala143 is represented by green side chain whereas the mutant residue, threonine is indicated by red side chain. This mutation is present on the surface of protein’s functional domain, glycoside hydrolase domain and is close to Cys142 that forms a disulfide bridge with Cys172. As a result of Ala143Thr variation, formation of Cys142-Cys172 disulfide bond results in local misconformation of protein. Moreover, the loss of Cys142-Cys172 disulfide bond can also affect the accessibility of nucleophilic residue, Asp170 in the active site. In addition, difference in physiochemical properties between wild-type and mutated residue can cause loss of interactions with the ligand, alpha-D-galactose thereby affecting the function of GLA.
History and clinical findings of mutation carriers
All family members positive for A143T/GLA (figure 1) had been healthy and did not use regular medications. The index patient, a female in her 60s, and her athletic son in his 30s were hospitalised at the Kuopio University Hospital because of several syncopes (table 1). They received dual-chamber pacemakers, the mother due to sick sinus syndrome and the son due to intermittent third degree atrioventricular block and asystoles up to 10 s, respectively. There was a suspicion of cardiac sarcoidosis in the elder son. He has been closely monitored with PET/CT but not treated with immunosuppressive medicines. The youngest son of the index patient in his 20s had also a syncope but no evidence of sick sinus syndrome or atrioventricular block. The mother suffered from tinnitus and somewhat poor heat tolerance, and her exercise capacity was limited. Few angiokeratomas were present in the typical paraumbilical area in the mother and the elder son. The other six family members with A143T/GLA had no FD-related symptoms or clinical findings. None of the A143T/GLA carriers had a history of hypertension, neurological problems or signs of kidney disease. All had normal creatinine and estimated glomerular filtration rate and no albuminuria (data not shown). In an ophthalmological examination, no cornea verticillata in the family members was identified. Brain MRI was available for the two sons of the index patient and was normal in both cases.
Biomarkers and the summary of cardiac findings of the patients with A143T/GLA and cardiomyopathy
In the spring 2015, both the index patient and the elder son were started with ERT (agalsidase-beta 1 mg/kg intravenous every other week). Tinnitus and poor heat tolerance that the mother used to have disappeared shortly thereafter and in 2 years her results in 6 min walking test normalised. Recently, she was diagnosed with mild hypertension and paroxysmal atrial fibrillation.
Biochemical biomarkers
Two sons of the index patient (table 1) and the school-aged daughter of the son in his 30s, all with A143T/GLA, had decreased levels of GLA activity in leucocytes. The elder son and his daughter had 25%, the younger son had <40% and the mother had 100% of the normal GLA activity, respectively. Lyso-Gb3 levels of the two sons were elevated in some but not in all measurements during a 3-year follow-up (table 1). Lyso-Gb3 levels in the index patient were in the upper normal range. The other relatives carrying A143T/GLA had normal GLA activities and lyso-Gb3 levels (data not shown).
The index patient had increased troponin T (TnT) and pro-B-type natriuretic peptide (proBNP) values (19 ng/L, reference value <15 ng/L; 1884 ng/L, reference value <900 ng/L, respectively) at the time of the diagnosis of cardiomyopathy. After starting ERT, all subsequent TnT and and proBNP values were normal. Both sons of the index patient and all other subjects with A143T/GLA had normal TnT and proBNP values (data not shown).
Standard 12-lead ECG
The ECG recording of the index patient showed sinus bradycardia, QRS complex width of 103 ms and 1 mm ST depression in anterolateral leads. The ECGs of both sons showed wide QRS complex (120 ms in the elder son and 105 ms in the younger son, respectively), and the elder son had prolonged PR time of 240 ms.
Cardiac imaging
The index patient and her two sons with A143T/GLA were diagnosed with cardiomyopathy in cardiac imaging. Their main FD-related cardiac imaging findings are shown in the table 1 and figures 3A–6B. Cardiac imaging findings in all other subjects with A143T/GLA were normal.
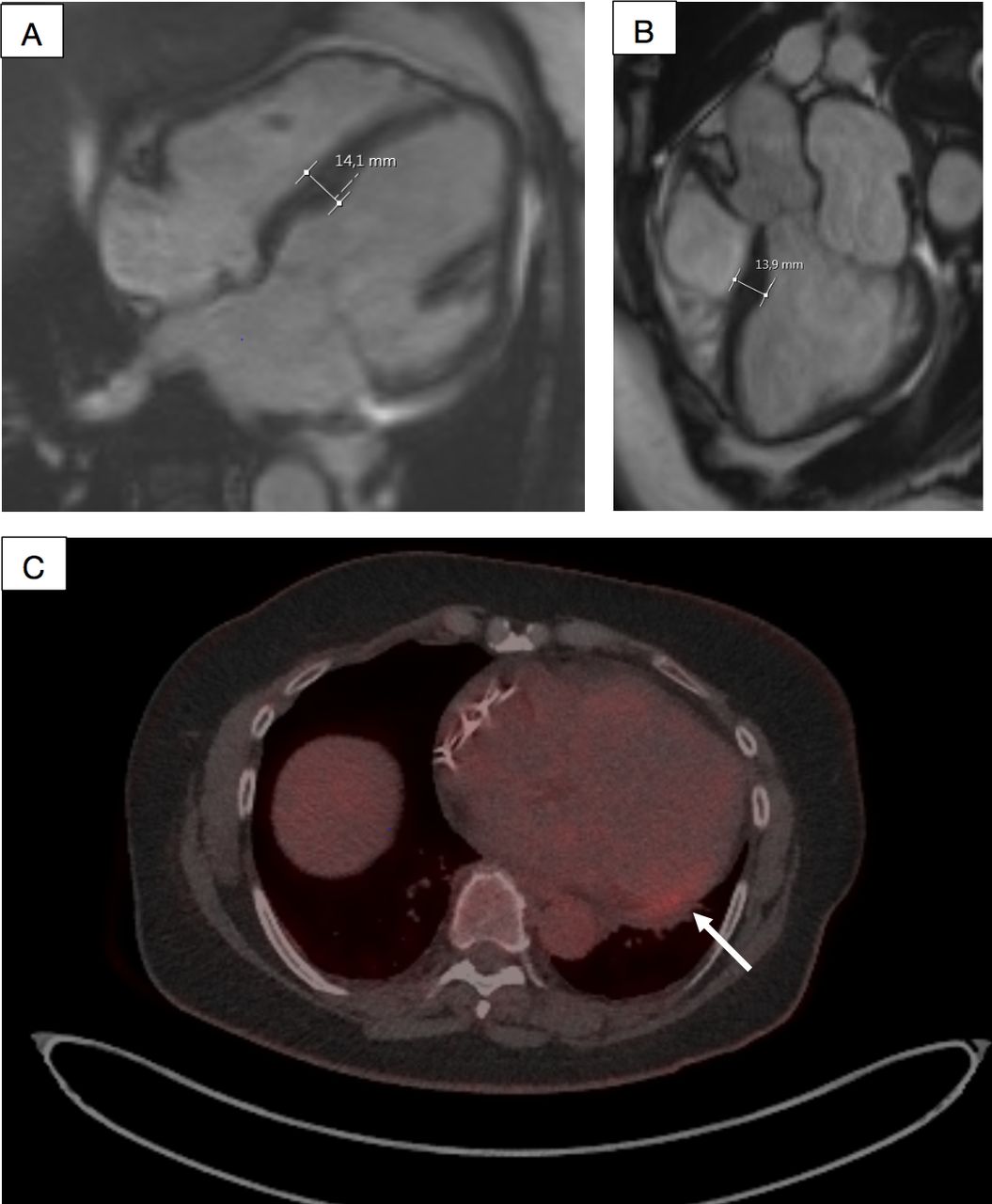
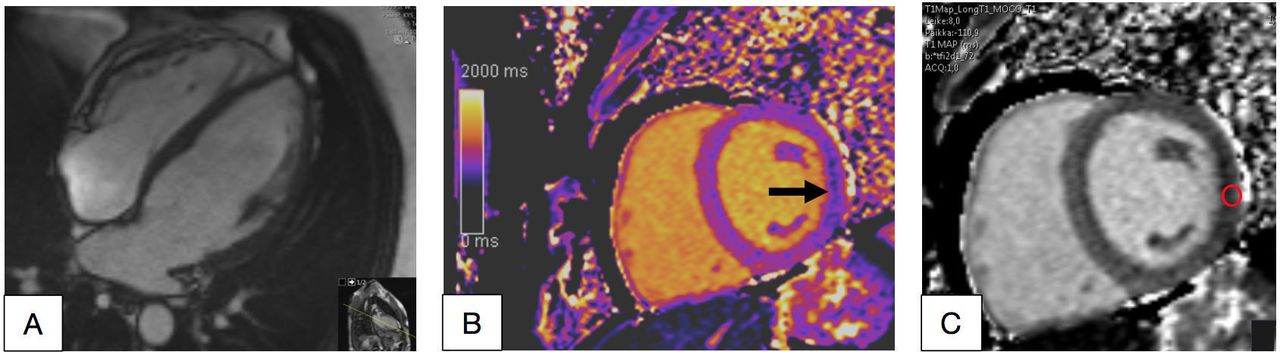
The index patient, a female in her 60s. Cardiac MRI showed signs of mild cardiomyopathy with the left ventricular (LV) maximal thickness of 14 mm in (A, B). 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT showed mild positive FDG signal (max standardised uptake value/lbm 3.1 and metabolic volume 0.4 mL) in PET images in the basal inferolateral wall of the LV (arrow) (C).

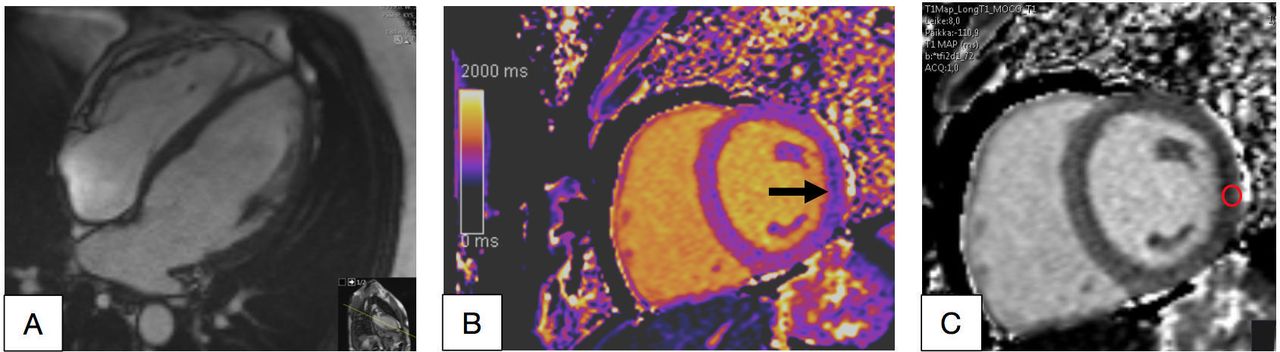
A male with cardiomyopathy in his 30s. In cardiac MRI, left ventricular (LV) was slightly enlarged (left ventricular end-diastolic volume index/end-systolic volume index 107/48 mL/m2) and the LV maximal thickness was 13 mm (A, B). Mild intramyocardial late gadolinium enhancement was seen in the typical basal inferolateral (BIFL) LV area (arrow, C, D). Increased T1 time 1113 ms (normal 950–1050 ms) was detected in the same BIFL segment (arrow and white circle).
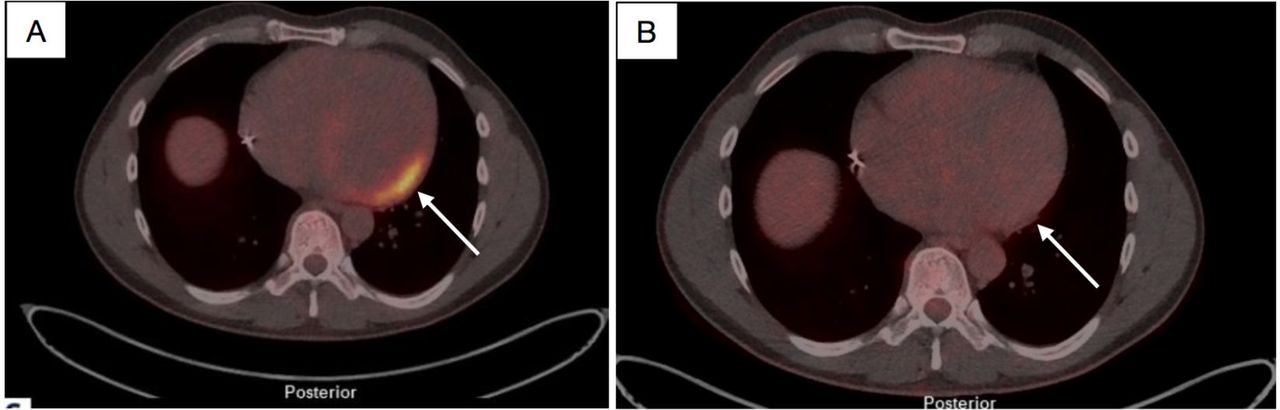
18F-fluorodeoxyglucose (FDG) positron emission tomography (PET)/CT pictures of the male in his 30s before and after he was treated with enzyme replacement therapy (ERT) for 1.5 years (A). Positive FDG signal (max standardised uptake value (SUV)/lbm 13.9 and metabolic volume 388 mL) was detected in PET images in the basal inferolateral wall of the left ventricular (arrow, B); 1.5 years after starting ERT, the FDG signal had almost disappeared (max SUV/lbm 2.3 and metabolic volume 0 mL).
Cardiac MRI of a male in his 20s with mild cardiomyopathy (A). Left ventricular (LV) was slightly enlarged (left ventricular end-diastolic volume index/end-systolic volume index 115/52 mL/m2) (B, C). T1 time was low 857 ms in the basal inferolateral wall of the LV (arrow and red circle).
The echocardiography and CMR of the index patient showed mild LV hypertrophy with the maximal thickness of 14 mm (figure 3A,B). PET/CT showed abnormal FDG uptake in the basal inferolateral (BIFL) wall of the LV (figure 3C).
The echocardiography and CMR of the elder son with A143T/GLA showed mild LV hypertrophy and slightly dilated LV (figure 4A). In CMR, there was myocardial LGE in the BIFL wall of the LV (figure 4B). Increased T1 time was detected in the same area (figure 4C-D). His first PET/CT study showed increased metabolic activity in the BIFL wall (figure 5A). The fourth PET/CT was carried out when he had been on ERT for 1.5 years, and then there was only minimal abnormal activity left in the BIFL LV wall (figure 5B).
CMR of the younger son showed LV maximal wall thickness in the upper limit of normal, slightly enlarged LV and low T1 time in the BIFL wall of the LV (figure 6A–C).
Endomyocardial biopsy
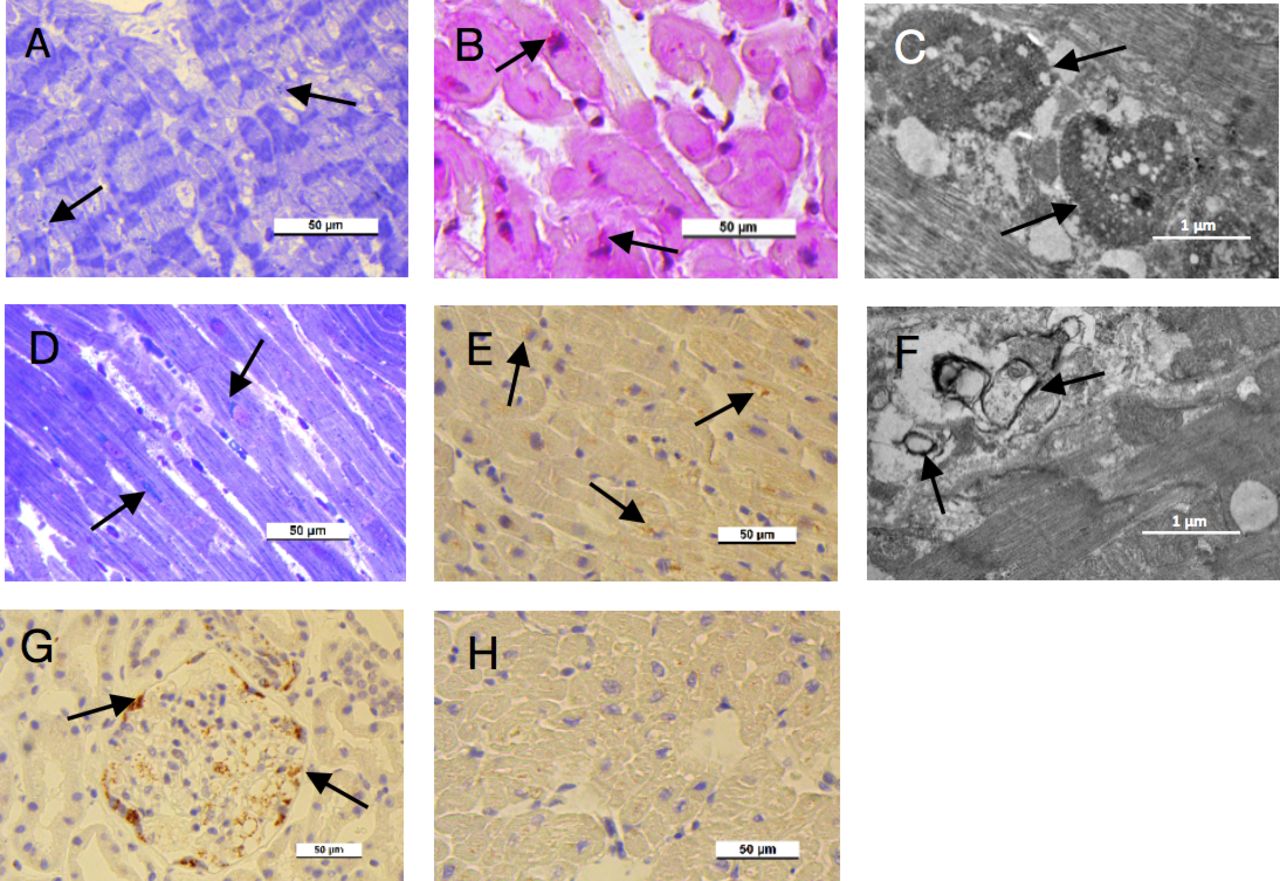
Endomyocardial biopsy of the sons of the index patient who were carriers of A143T/GLA showed evidence of glycolipid accumulation in cardiomyocytes (figure 7A–F). In light microscopy of the elder son, there were vacuoles in myocytes in toluidine blue staining, and accumulation of PAS (periodic-acid-Schiff) -positive material, compatible with Gb3 deposits typical of Fabry cardiomyopathy (figure 7). Cardiomyocytes of the younger son showed abnormal accumulations in toluidine blue stain, which stained with an anti-Gb antibody, suggesting Fabry cardiomyopathy (figure 7D,E). Renal cells in kidney specimen of a patient with confirmed FD caused by the classical mutation GLA-Arg220Ter stained with anti-Gb3 antibody (figure 7G). Myocardium specimen of a control cadaver heart showed no Gb3 positivity (figure 7H).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A–C) Endomyocardial specimens from the elder son. (A) Accumulations (arrows) and vacuoles in the cardiac cells, light microscopy, toluidine blue stain. (B) PAS-positive material (arrows) in cardiac cells, light microscopy, PAS stain. (C) Lysosomal inclusions (arrows) inside the cardiac cell, an electron micrograph. (D–F) Endomyocardial specimens from the younger son. (D) Accumulations (arrows) in the cardiac cells, light microscopy, toluidine blue stain. (E) Globotriaosylceramide (Gb3)-positive material (arrows) in cardiac cells, light microscopy, Gb3 immunohistochemical stain. (F) Lamellar deposits (arrows) inside the cardiac cell, an electron micrograph. (G, H) Control stains. (G) Gb3-positive material (arrows) in a kidney specimen of a female patient in her 40s with Fabry disease caused by the classical mutation GLA-Arg220Ter. (H) Negative Gb3 staining in normal cardiac cells of a control cadaver.
In electron microscopy, cardiomyocytes of the hemizygous sons showed lysosomal inclusions and lamellar deposits, respectively, compatible with lysosomal Gb3 accumulation (figure 7C and F).
Discussion
Principal findings
In the present study, we describe a Finnish family with A143T/GLA, in which the index female and both adult males carrying the variant had cardiomyopathy compatible with Fabry cardiomyopathy in the absence of renal or neurological manifestations. Our study suggests that A143T/GLA is a late-onset FD-causing variant with incomplete penetrance and predominantly cardiac manifestations based on the following findings. First, we diagnosed familial cardiomyopathy with apparent X linked inheritance and age-related penetrance, and earlier onset in males, all features compatible with Fabry cardiomyopathy. Young females with the variant had no cardiomyopathy, which is typical for late-onset FD. Second, no pathogenic variants except for A143T/GLA was identified in the screening of 59 cardiomyopathy-related genes in the index patient and his two sons with cardiomyopathy. Third, symptoms and clinical findings in the proband and her two sons with A143T/GLA, including atrioventricular conduction defect, sick sinus syndrome, arrhythmias and angiokeratoma, are typical for patients with late-onset FD. Fourth, biomarkers in patients with cardiomyopathy suggest late-onset FD. In hemizygous males with the mutation, levels of GLA enzyme activity were below or in the borderline area of the diagnostic value of <35%.20 GLA activity was low also in the young granddaughter of the index patient. GLA activity in the index patient was normal, but normal GLA activity does not rule out FD in females. Lyso-Gb3 levels were elevated at least in one measurement in both males, and in the upper normal range in the mother. Fifth, cardiac imaging in the proband and two hemizygous sons showed variable combinations of cardiomyopathy, increased metabolic activity in the PET/CT and late enhancement in the BIFL area of LV in CMR, and abnormally low or high myocardial T1 mapping values, all features very suggestive of Fabry cardiomyopathy. Sixth, in the endomyocardial biopsies of the hemizygous males, vacuoles and accumulation of abnormal PAS-positive material in the myocytes were found in the light microscopic examination. The electron microscopic examination showed abnormal deposits and/or lamellar structures in the cytoplasm of myocytes, compatible with Gb3 deposits. Finally, myocytes stained with an anti-Gb3 antibody were available for the younger son.
In the context of current literature
With increasing use of genetic testing in patients with cardiomyopathy, variants with uncertain clinical significance are discovered more frequently. One challenge for clinicians as well as anxiety for patients is whether A143T/GLA is a FD-causing mutation or not.8 The first Fabry case described by Anderson in 1898 had A143T/GLA.9 Since then, A143T/GLA has been found in variable numbers in general populations, as well as in hypertrophic cardiomyopathy and Fabry patient populations. According to the latest Genome Aggregation Database, the allele frequency of A143T/GLA in 205 433 alleles in non-selected subjects is 5.06e-4. In newborn screening in the USA, the prevalence of A143T/GLA was 1:3800 in some areas.7 Newborn screening of 37 104 males in Piemonte, Italy revealed three males with A143T/GLA.10 In our study of 382 Finnish patients with hypertrophic cardiomyopathy, A143T/GLA was found in two patients.21 In a recent abstract of the Fabry Outcome Survey, the prevalence of A143T/GLA was 3.7% (60 of 1602 patients with FD), making it the fourth most common genetic variant in the patients with FD in the Fabry Outcome Survey.11
Is A143T/GLA a FD-causing variant? The first FD case described by Anderson had A143T/GLA and classic FD, but the relatives with A143T/GLA showed a substantial variation in phenotypic expression of the disease through multiple generations.9 Since 2013, seven articles with a total of 88 adults with A143T/GLA have been published. In these studies, the phenotype varied from the classic FD to healthy unaffected patients with normal GLA enzyme activities, resulting in contradictory interpretation of the pathogenicity of the variant.12–18 According to the latest ClinVar variant classification, A143T/GLA has conflicting interpretations of pathogenicity.
The main clinical features of Fabry cardiomyopathy are progressive LV hypertrophy resulting in heart failure, usually mild valvular heart disease, conduction abnormalities, supraventricular and ventricular arrhythmias and sudden death. Within the heart, glycolipids accumulate in several cell types including cardiomyocytes and conduction system cells.1 2 Lipid accumulation leads to inflammation and fibrosis, which finally results in irreversible tissue damage. In CMR, Fabry cardiomyopathy is characterised by myocardial LGE in the BIFL wall of the LV and a reduction in non-contrast T1 signal.22 23 Low (<900 ms) T1 mapping value indicates early cardiac lipid accumulation and is present before LVH and fibrosis develop.24 When permanent damage ensues, T1 signals rise and T1 mapping loses its value as a reliable tool for Fabry cardiomyopathy diagnosis.25 Endomyocardial biopsy should be considered, when adults with cardiomyopathy have GLA variants of unknown significance.26 Lipid accumulation, hypertrophy and vacuoles in cardiomyocytes, lysosomal inclusions, Gb3-positive immunohistochemistry and typical lamellar deposits on electron microscopy characterise Fabry cardiomyopathy.2 In the present study, subjects with A143T/GLA and cardiomyopathy had imaging and histological features highly suggestive of Fabry cardiomyopathy. Our study supports the notion that A143T/GLA is a late-onset FD-causing variant with incomplete gender-related and age-related penetrance.
Possible mechanisms
Late-onset FD, mainly caused by missense mutations in GLA, is characterised by residual enzyme activity.1 6 Cardiomyopathy is often the predominant, or the only manifestation of the late-onset disease, and typically develops in middle-aged hemizygous and heterozygous subjects.1 On the other hand, not all females with pathogenic GLA mutations will develop clinical FD, possibly because of skewed X chromosome inactivation.2 27 Consequently, it may be difficult to define pathogenicity of a late-onset GLA variant, such as A143T/GLA, particularly if the family with the mutation is small, its members are young and/or females and no CMR has been used to study the presence of subtle cardiomyopathy. Furthermore, there is recent evidence that rather than pure storage disease, Fabry cardiomyopathy may be a chronic inflammatory cardiomyopathy triggered by sphingolipid accumulation.28–30 18F-FDG uptake representing inflammation appears to precede the development of myocardial fibrosis.30 Consequently, the degree of inflammatory response might affect the onset and severity of cardiomyopathy in late-onset mutation carriers with residual enzyme activity and moderate burden of lipid accumulation.
Clinical implications
We suggest that patients carrying the A143T/GLA mutation should be carefully examined and followed by a cardiologist familiar with Fabry cardiomyopathy. CMR, especially non-contrast T1 mapping is an important diagnostic study for subjects carrying the mutation. PET/CT may bring added value to the diagnostics, treatment decisions and follow-up, particularly if CMR is contraindicated, T1 mapping is not available or CMR findings are not unequivocal.
Strengths and limitations
Even if we had a limited number of subjects in the present study, they were examined comprehensively with targeted large-scale next-generation genetic panels, biomarkers, extensive cardiac imaging and endomyocardial biopsies.
Conclusions
A143T/GLA is very likely a late-onset FD-causing variant with incomplete age-related and gender-related penetrance and predominantly cardiac manifestations. CMR, including T1 mapping and PET/CT appear to be useful in diagnosis, treatment decisions and the follow-up of subjects suspected of having Fabry cardiomyopathy.
Key messages
What is already known on this subject?
Late-onset Fabry disease (FD) is often associated with cardiac manifestations.
If not treated early enough, Fabry cardiomyopathy considerably affects the quality of life and shortens life expectancy, necessitating timely diagnosis.
Even if Ala143Thr variant of the α-galactosidase A gene (A143T/GLA) is one of the most common GLA variants, there is conflicting evidence on its pathogenicity.
What might this study add?
The A143T/GLA is associated with late-onset cardiomyopathy with incomplete penetrance and predominantly cardiac manifestations.
Cardiac magnetic resonance (CMR) imaging with T1 mapping and PET/CT imaging bring added value to the treatment decisions and the follow-up of late-onset FD.
How might this impact on clinical practice?
Patients carrying the A143T/GLA mutation should be carefully examined and followed by a cardiologist familiar with FD.
CMR, especially non-contrast T1 mapping, and in unequivocal cases, 18F-fluorodeoxyglucose positron emission tomography/CT should be used to detect cardiomyopathy.
Supplemental material
Acknowledgments
The authors would like to thank all participating hospitals and care providers for their co-operation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KV: study, analysis, reporting, clinical examination, adult echo, writing manuscript. JN-Q, TL, MH and L-LR: imaging, analysis. AN: histological analysis. ML and MM: genetic analyses. JK: study conception, analysis, writing manuscript. IK: study conception, writing and revision of manuscript.
Funding The study was supported by Sanofi-Genzyme (grant to KV and JK) and by the Academy of Finland, the Finnish Heart Research Foundation and the Kuopio University Hospital (grants to JK).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The study protocol was approved by the Ethics Committee of the University Hospital of Kuopio and was performed in accordance with the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.